 2019, Vol. 30
2019, Vol. 30 


b Department of Chemistry, Bengbu Medical College, Bengbu 233030, China

Prof. Lijuan Jiao received her Bachelor’s degree (2000) from Shandong University, China, and obtained her Master’s degree (2003) under the supervision of Prof. Guanwu Wang at University of Science and Technology of China (USTC). She then moved to Louisiana State University, U. S. A., and obtained her Ph.D. (2007) under the supervision of Prof. Kevin M. Smith. She joined Anhui Normal University in 2008, and became a full professor (2010) at College of Chemistry and Materials Science. Her researchfocuses on the developmentof novel BODIPYand porphyrin related dyes, understanding their photophysical properties and studying their optoelectrical and biological applications. She received a SPP/JPP Young Investigator Award for her research in BODIPY chemistry in 2016.
Small-molecule fluorophores are important tools to investigate biological processes and are prevalent in many modern applications, including bioimaging, sensing, theranostic, and optoelectronic materials [1]. Recently, BODIPY (4, 4'-difluoro-4-bora-3a, 4adiaza-s-indacene) dyes [2-4], the boron complexed dipyrromethenes, have been found wide applications in various research areas since discovered in 1968 by Treibs and Kreuzer [5]. The growing success of these scaffolds gives the credit to their remarkable photophysical properties such as large molar absorptivity, sharp fluorescence emissions, high fluorescence quantum yields, and high photostability [6]. Moreover, these diverse applications thus further demand highly structure-diversified BODIPY dyes with various tunable photophysical properties.
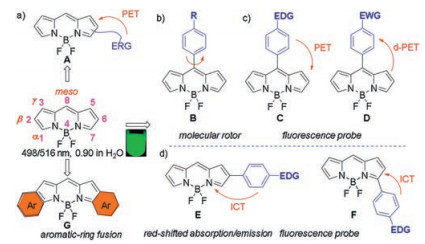
The typical UV–vis absorption spectrum of BODIPY framework is mainly located in the short-wave region (470–530 nm) (Fig. 1). For example, the parent unsubstituted BODIPY absorbs at 498 nm with an emission maximum at 519 nm in water. It also has a high fluorescent quantum yield of 0.90 in water (Fig. 1) [7]. It has been found that various structure modifications on BODIPYs can finetune their photophysical properties. Attaching electron rich groups (for example, N and S containing groups) often gives BODIPYs with decreased fluorescence (A, Fig. 1) through photo-induced electron transfer (PET) process [8]. After binding with metal ions or changing pH, this PET process can be blocked to restore their strong fluorescence. Similarly, meso-aryl groups with electron donating groups (C, Fig. 1) [9] or electron withdrawing groups (D, Fig. 1) [10] have been widely used to tune the photophysical properties through PET process or d-PET process, respectively.

|
Download:
|
| Fig. 1. (a) The core structure of BODIPY dyes and some representative methods for tuning their photophysical properties; (b) BODIPY molecular rotor model; (c) BODIPY fluorescence probe model according to PET process and d-PET process; (d) BODIPY red-shifted strategy and fluorescence probe according to ICT process. ERG: electron rich group; EDG: electron donor group; EWG: electron withdrawn group. | |
{kind=link}
Moreover, various meso-arylBODIPYs B (Fig. 1) with no substituents on the 1, 7-positions have been developed as efficient fluorescent molecular rotors (FMR) [11]. The efficient strategy for these BODIPY FMRs is based on the rotation of the meso-phenyl group around the C—C single bond, which causes a non-radiative decay at the excited state in non-viscous media. The increase of viscosity makes it difficult for the meso-phenyl group to rotate and hence inhabits the non-radiative decay process and brings the increase in the fluorescence intensity and fluorescence lifetime.
Some applications such as biological imaging generally require dyes that absorb and emit at longer wavelengths, in the far-red or NIR region [12]. To realize this objective, various structural modifications can also be performed on the BODIPY skeleton [2c, 13]. Attaching aromatic substituents at α or β positions can extend π-conjugations and give red-shifted BODIPY derivatives. Especially, BODIPYs E and F (Fig. 1) with electron donor groups have large red-shifted absorption and emission maximum due to the presence of intramolecular charge transfer (ICT) process [14]. The presence of ICT process will also decrease the fluorescence quantum yields of these derivatives especially in polar solvents. Blocking the ICT process often gives enhanced fluorescence with ratiometric changes. On the other hand, π-extension by fusing aromatic rings to BODIPY core is particularly promising to generate required dyes that absorb and emit at longer wavelengths, and has resulted many elegant new aromatic ring-fused BODIPY derivatives (For example G in Fig. 1) [15].
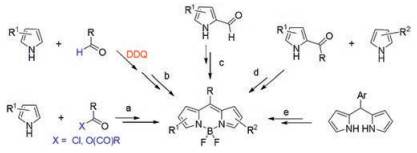
2. Overview of the traditional syntheses of BODIPY derivativesBecause of the structural characteristics of the BODIPY molecule itself, two main paths can be typically adopted to obtain functionalized BODIPY derivatives. The first one is the traditional functionalization of BODIPY dyes achieved by de novo synthesis using suitably prefunctionalized pyrroles (Scheme 1). The first one-pot approach for symmetrical BODIPYs is an acid-catalyzed condensation between pyrrole and acid chloride or anhydride, followed by in situ boron complexation using NEt3 and BF3·Et2O (Scheme 1, route a) [16]. Similarly, acid-catalyzed condensation of pyrrole with an aldehyde (typical aromatic aldehyde, excess amount) in the presence of an oxidant (DDQ, for example) has also been widely used (Scheme 1, route b) [17]. An interesting alternative to synthesize symmetrical BODIPY derivatives without meso-substituents has been developed through self-condensation of pyrrole-2-carbaldehyde derivatives in the presence of phosphorous oxytrichloride (POCl3) (Scheme 1, route c) [18]. Furthermore, two different pyrrole moieties can also be used in this condensation reaction, providing various unsymmetrical BODIPYs in onepot (Scheme 1, route d) [19]. Finally, a popular approach to construct meso-arylBODIPYs without pyrrolic substituents starts from the oxidation of dipyrromethanes, followed by boron complexation through an excess of base and BF3.OEt2 (Scheme 1, route e) [20]. The corresponding aryl containing dipyrromethane, which is relatively stable, now can be efficiently synthesized through a convenient, HCl-catalyzed condensation between aldehyde and pyrrole in water [21]. Precipitation of the formed dipyrromethane from the aqueous solution is the key to avoid polymerization and to isolate the target compound.

|
Download:
|
| Scheme 1. Representative de nova syntheses of BODIPY dyes. | |
{kind=link}
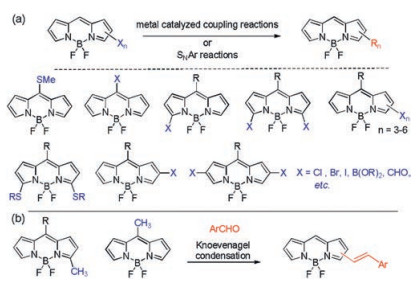
The other approach is the postfunctionalization of BODIPY dyes, for example, by conversion of halogen, methyl or thioether installed on the BODIPY fluorophores (Scheme 2) [22]. The postfunctionalization strategy avoids the tedious synthesis of unstable pyrrole derivatives and is thus more attractive. The most common reactive functional group attached to the BODIPY core structure for postfunctionalization is a halogen [23]. Halogenated BODIPYs were synthesized through de novo synthesis using halogenated pyrroles or dipyrromethanes as building blocks, electrophilic halogenation of BODIPYs and α-chlorination of BODIPYs with CuCl2. Meso- and α-thioether containing BODIPYs were traditionally synthesized through de novo synthesis [24] or nucleophilic substitution (SNAr) reactions of the corresponding halogenated BODIPYs [25]. These reactive BODIPY dyes have been widely used to synthesize diverse functionalized derivatives through SNAr reactions [26] as well as transition-metal-catalyzed C—C coupling reactions (Scheme 2a) [13, 27]. Methyl groups directly attached to the BODIPY core at α- or meso- positions are relatively acidic and are reactive functional sites via the wellknown Knoevenagel reaction (Scheme 2b), forming alkenyl BODIPY derivatives from aromatic aldehydes [28]. Various formyl BODIPY derivatives, as a class of important reactive BODIPY dyes for further transformation were also synthesized through de novo synthesis [21, 29] or oxidation of the corresponding methyl group [30].

|
Download:
|
| Scheme 2. Representative functionalization of BODIPY dyes via metal catalyzed coupling reactions and Knoevenagel condensations. | |
{kind=link}
In recent years, rapid and site-selective functionalization of aromatic systems by direct C–H bond activation has emerged as a convenient approach for preparing advanced organic molecules. These above described functionalization methods, although elegant, often require the preparation of reactive BODIPY dyes from unstable intermediates and/or a long synthetic route. To address these issues, in the last few years, growing research efforts have been devoted to the direct C–H functionalization of the BODIPY core, because it allows the facile installation of desired functional groups in a single atom economical step. In this review, we will focus on the summary of the direct C–H modification of the BODIPY core on the pyrrolic ring developed in recent years. We mainly discuss the synthesis method and simply summarize the properties and applications of some compounds involved.
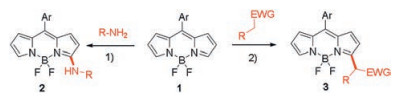
3. Oxidative nucleophilic hydrogen substitution reactionInterconversion of halogen group containing BODIPY by nucleophilic reagent is a textbook SNAr reaction. However, BODIPY without halogen or any other leaving group can also be directly functionalized by various nucleophiles. Indeed, the pyrrolicunsubstituted BODIPY dyes are highly reactive toward oxidative nucleophilic substitution of the α-hydrogen atom(s). Dehaen's group found BODIPY 1 can be directly modified with nucleophilic reagents on the 3(, 5)-positions in the presence of oxygen (Scheme 3) [31]. The nucleophiles are limited to aliphatic amines (products 2) and carbanions (products 3) in order to give good yields. Both primary and secondary aliphatic amines readily participate in this oxidative nucleophilic substitution of hydrogen, yielding 3-substituted BODIPYs, while no disubstituted product is formed.

|
Download:
|
| Scheme 3. Direct functionalization of the BODIPY core at the 3(, 5)-positions by oxidative nucleophilic substitution of hydrogen.1) O2, DMF, room temp. 2) O2, DMF, base, r.t. | |
{kind=link}
This reactionwas further developed without oxidant conditions [32]. If the nucleophile contains a leaving group, the hydrogen of BODIPY 1 can be replaced in the oxidizer-free condition, to give compound 4 (Scheme 4). This methodology has also been combined with a reversible Michael addition on nitrostyrenes to provide a novel, highly efficient access to the valuable 3-styrylated BODIPY dyes 5 (Scheme 4).

|
Download:
|
| Scheme 4. Vicarious nucleophilic substitution and the reversible Michael addition on nitrostyrenes in tandem with vicarious nucleophilic substitution of the 3-hydrogen of BODIPY 1. 1) DMF, r.t., base. 2) DMF, r.t., PhSH (10 mol%) as activating nucleophilic organocatalyst, 18-crown-6 (cat.), K2CO3. | |
{kind=link}
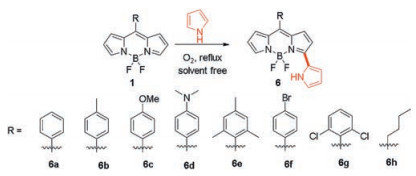
The installation of aromatic moieties, including aryl units onto the 3(, 5)-positions is a common strategy to extend the conjugation of the BODIPY chromophore to achieve BODIPY dyes with longwavelength absorption and emission. α-ArylBODIPYs are traditionally synthesized via de novo synthesis from suitable α-arylpyrroles or the metal-catalyzed coupling reactions on α-haloBODIPYs. The later one requires the installation of the halogen atoms which are subsequently replaced by aryl moieties. In 2012, Jiao and coworkers showed that the 3-hydrogen of BODIPY dyes 1 can be directly oxidative nucleophilic substituted by pyrrole under reflux condition under oxygen atmosphere, from which a series of pyrrolyldipyrrinato BF2 complexes 6a–h (Scheme 5), as extended BODIPYs, were synthesized [33]. Most of these BODIPYs show strong fluorescence emissions at wavelengths over 600 nm in six solvents of different polarity, while only 6d has very weak fluorescence due to the PET process from the electron-rich mesoaminophenyl group to the BODIPY core.

|
Download:
|
| Scheme 5. Syntheses of BODIPYs 6 via solvent free oxidative nucleophilic substitution. | |
{kind=link}
4. Free radical reaction 4.1. The C—C bond formation
Due to the limited reactivities of most available nucleophiles, the use of the above nucleophilic reactions to develop 3, 5-diarylBODIPY is greatly limited. Recently, free radical reactions are widely used in organic synthesis because they have high reactivity and can break the inert C–H bond. In 2015, Dehaen and co-workers have made an elegant discovery towards the regioselective α-arylation of the BODIPY system. They reported a very useful radical reaction on BODIPY via the ferrocene-mediated radical arylation using aryldiazonium salts (Scheme 6) [34].

|
Download:
|
| Scheme 6. Radical C–H diarylation of 3, 5-unsubstituted BODIPY dyes 7 with excess of various aryldiazonium salts. Ar=2, 6-dichlorophenyl (unless stated otherwise).1) 1 equiv. aryldiazonium tetrafluoroborate, ferrocene, acetone, r.t. 2) 2.5 equiv. aryldiazonium tetrafluoroborate, ferrocene, acetone, r.t. | |
{kind=link}
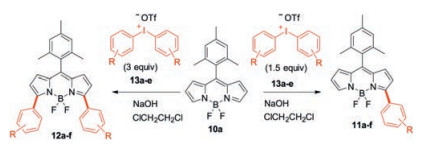
Diaryliodonium salts feature with easy accessibility, nontoxicity, good stability (bench stable) and high reactivity at suitable conditions. Jiao and co-workers recently found that diaryliodonium salts are suitable arylation reagent for the regioselective α-arylation of BODIPY [35]. In the presence of 1.5 equiv. of diaryliodonium salts, the regioselective α-arylation of BODIPY 10a smoothly proceeded with the assistance of NaOH to generate exclusively α-arylBODIPYs 11 (Scheme 7). Further increasing the amount of diaryliodonium salts to 3.0 equiv., the corresponding 3, 5-diarylBODIPYs 12 were obtained. This metalfree and α-selective arylation of BODIPYs provides a straightforward access to a variety of α-arylBODIPY dyes. It comes through a radical reaction pathway, involving the in situ generation of aryl radical species from the decomposition of diaryliodonium salts. These aryl radical species then react with BODIPYat the α-position.

|
Download:
|
| Scheme 7. Synthesis of 3(, 5)-diarylBODIPYs 11 and 12 from the reaction of α-free BODIPYs with diaryliodonium salts 13a-e. | |
{kind=link}
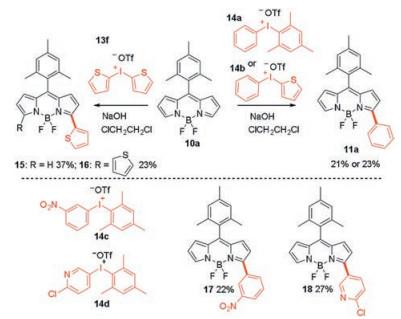
This direct α-arylation reaction is highly compatible with various functionalities. Besides diaryliodonium salts, other heteroaromatic hypervalent iodium salts, like di(thiophene-2-yl)iodonium salt, are also suitable for this reaction. The resulting dyes showed strong absorption and emission over a broad range of spectra tunable via the simple variation of the diaryliodonium salts. Interestingly, a high chemoselectivity is observed for this α-arylation reaction in case of unsymmetrical diaryliodonium salts. For example, the reaction of diaryliodonium salts 14a and 14b with BODIPY 10a, the less bulky phenyl moiety was installed onto the α-position of the BODIPY core (Scheme 8). Similar result was observed when 14c and 14d were used as the arylation agents.

|
Download:
|
| Scheme 8. Regioselective α-arylation of BODIPY 10a with 13f and unsymmetrical diaryliodonium salts 14a-d. | |
{kind=link}
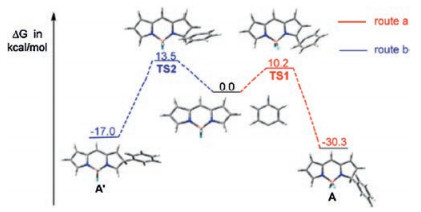
Furthermore, a computational study at the B3LYP/6–31 G(d) level using the Gaussian 09 program was performed to understand the reason for the regioselective formation of α-arylBODIPY instead of β-arylBODIPY (Fig. 2). The formation of the transition state TS2 (route b) requires a higher activation energy than that required for the formation of transition state TS1 (route a).

|
Download:
|
| Fig. 2. Free energy profiles for the two competitive radical reaction pathways in the radical reaction of BODIPY 10 and the in situ generated phenyl radical at 298.15 K: Structures of transition states TS1 (route a) and TS2 (route b), structures of the intermediates A (route a) and A' (route b), and their corresponding activation free energies. Reproduced with permission [35]. Copyright 2016, American Chemical Society. | |
{kind=link}
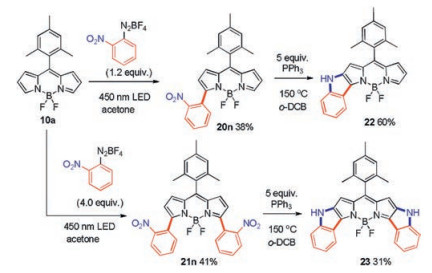
Using a similar approach, other free radical generating methods are also used to introduce aryl groups on the BODIPY backbone. In 2019, Jiao's group successfully prepared 3(, 5)-(di)arylBODIPYs using BODIPY 19 and aryl diazonium salt, under the irradiation of visible light in the absence of any external photoredox catalyst (Scheme 9) [36]. The chemistry is driven by the singlet excited state (1PS*) of BODIPYs upon visible-light absorption while successively triggering the formation of aryl radicals from aryl diazonium salts. It is only necessary to control the equiv of the aryl diazonium salts to obtain the 3-arylBODIPY 20 or the 3, 5-diarylBODIPY 21, respectively.

|
Download:
|
| Scheme 9. The photo-induced synthesis of BODIPYs 20 and 21. | |
{kind=link}
Using this convenient, photo-induced C–H arylation, BODIPYs 20n and 21n with ortho-nitro groups on the 3(, 5)-(di)phenyl substituents were obtained in 38% and 41% yields, respectively. Next, BODIPYs 20n and 21n were smoothly transformed into indole-fused BODIPYs 22 and 23 in 60% and 31% yields (Scheme 10), respectively, via an intramolecular reductive cyclization (the Cadogan reaction). These ring fused compounds show remarkably red-shifted absorption and emission spectra. In dichloromethane, mono-indole-fused BODIPY 22 has a broad absorption spectrum with a maximum absorption peak at 596 nm, whereas diindoleannulated BODIPY 23 shows a more than 84 nm further bathochromic shift to 680 nm. The maximum peak of emission spectrum for both compounds reach 632 nm and 701 nm, respectively. Due to its NIR absorption and bright fluorescence, 23, with a fluorescence quantum yield of 0.31 in dichloromethane, was further evaluated for fluorescence imaging in HeLa cells, which was readily taken up by cells in only 15 min and showed bright red fluorescence in the cytoplasm (Fig. 3). Furthermore, there is no evident cell death even at a concentration of 50 μmol/L by the CCK-8 assay, indicating the good biocompatibility of 23 to cells.

|
Download:
|
| Scheme 10. Synthesis of BODIPYs 22 and 23. | |
{kind=link}

|
Download:
|
| Fig. 3. Confocal fluorescence images of HeLa cells stained with 23 (5 μmol/L) and DAPI. (a) DAPI fluorescence and (b) 23 fluorescence after incubation for 15 min. (c) Merged images of parts b and c. Copied with permission [36]. Copyright 2019, American Chemical Society. | |
{kind=link}
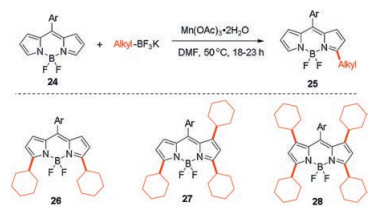
Not only aryl radicals, but also alkyl radicals can also achieve similar results [37, 38]. In 2015, Dehaen's group successfully synthesized alkylated BODIPYs using free radical C–H alkylation based on the oxidation of alkyl boronic acids or potassium trifluoroborates with manganese(Ⅲ) acetate [37]. The α-alkylBODIPYs 25 were obtained under 1.0 equiv. of various alkyl potassium trifluoroborates and 2.5 equiv. of manganese(Ⅲ) acetate (Scheme 11). The 3, 5-di-, 1, 3, 5-tri-, and 1, 3, 5, 7-tetraalkylated fluorophores 26-28 can also be prepared in good yields by increasing the amount of alkyl potassium trifluoroborates and manganese(Ⅲ) acetate. These BODIPYs with multiple bulky cycloalkyl groups exhibit good solid-state emission.

|
Download:
|
| Scheme 11. Regioselective synthesis of mono-, di-, tri-, and tetraalkylated BODIPYs. | |
{kind=link}
Alkylated BODIPYs can also be successfully synthesized from BODIPYs and alkyl diacyl peroxides as recently reported by Jiao's group in 2019 [38]. Compound 31 was prepared from BODIPY 29 with 1 equiv. diacyl peroxide (Scheme 12). Cu(acac)2 as the catalyst in PhCl at 90℃ C for 5h gave the best results. Dialkylated compound 32 was also obtained under the same condition only by increasing the amount of peroxides to 2 equiv. This highly efficient method exhibits excellent chemoselectivity, affording a broad range of structurally diverse and exclusively α-alkylated BODIPYs in the presence of catalytic Cu(acac)2 via radical process. The versatile functional groups of these alkylated BODIPY fluorophores (for example, BODIPYs 31a-c) render further useful transformation, labeling and tethering of molecules and biomacromolecules of interest.

|
Download:
|
| Scheme 12. Synthesis of 3(, 5)-(di)alkylated BODIPYs 31 and 32. | |
{kind=link}
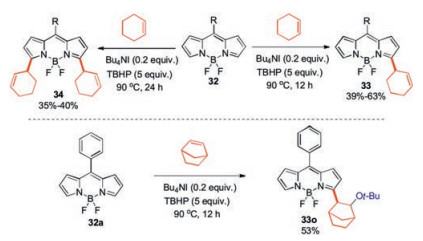
Among the above synthetic methods, the free radical species are often obtained by oxidation, reduction, or metal catalysis from the prefunctionalized precursors. Very recently, Jiao and coworkers have developed a Bu4NI/tBuOOH-catalyzed, highly regioselective cross-dehydrogenative coupling reaction (CDC) of the α-C–H bond(s) of the BODIPY core [39]. This reaction utilizes various commercial allylic alkenes, and provides an efficient route to α-functionalized BODIPYs that were previously difficult to access. The α-regioselective alkylation reaction was believed to involve a radical process through hydrogen atom transfer process from allylic alkene to tert-butoxy radical initiated by the Bu4NI/TBHP catalytic system. In the presence of 0.2 equiv. of Bu4NI and 5 equiv. of TBHP at 90℃, major products 33 were isolated in 39%-63% yields after 12h. By simplyextending the reaction time to 24 h, the corresponding diallylic alkylation products 34 were regioselectively produced in 35%–40% yields (Scheme 13).

|
Download:
|
| Scheme 13. Bu4NI/TBHP catalyzed regioselective mono- and diallylic alkylation of BODIPYs 32 at α-position(s) with alkenes. | |
{kind=link}
Interestingly, the reaction of BODIPY 32a with norbornene possessing two tertiary allylic carbons afforded BODIPY 33o in 53% yield. This may be attributed to the high reactivity of the double bond in norbornene, involving the addition of the tert-butoxy radical to norbornene first [39].
This high α-regioselectivity of this CDC reaction is also suited to introduce ethers onto the BODIPY chromophore. To illustrate the wide applicability of this regioselective reaction, the authors further extended the reaction of BODIPY 32 with a set of common ethers (Scheme 14). For example, BODIPY 32a reacted smoothly with diethyl ether and dibutyl ether, producing the corresponding α-alkylBODIPYs 35a and 35b in 50% and 52% isolated yields, respectively. In addition, common cyclic ethers, such as tetrahydrofuran and 1, 4-dioxane, also reacted efficiently with BODIPY 32a under the optimized reaction conditions, from which the desired α-functionalized products 35c and 35d were obtained in 55% and 42% isolated yields, respectively. This regioselective alkylation reaction was also used in the installation of 1, 4, 7, 10, 13, 16-hexaoxacyclooctadecane (18-crown-6) onto the BODIPY chromophore. Compound 32a reacted smoothly with 18-crown-6 to give BODIPY 35e in a 43% isolated yield.

|
Download:
|
| Scheme 14. Synthesis of BODIPYs 35a-e from BODIPY 32a with ethers. | |
{kind=link}
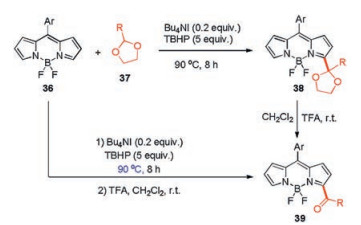
Using a similar reaction, the acetal/ketal can be introduced at the 3(, 5)-position of BODIPY to give compound 38. Furthermore, 3(, 5)-acylated BODIPY 39 was obtained by hydrolysis of 38 under acidic condition or in a one-pot reaction from BODIPY 36 (Scheme 15) [40].

|
Download:
|
| Scheme 15. One-pot and step by step synthesis of α-acylated BODIPYs 39. | |
{kind=link}
In 2018, an oxidative cross-dehydrogenative coupling of BODIPYs with toluene and its derivatives has been developed by Jiao's group [41]. This method exhibits excellent chemoselectivity, affordingexclusively α-benzylated BODIPYs 42 in the presence of t-BuOOH and a catalytic amount of Cu(OAc)2. Interestingly, by simply increasing the amount of TBHP to 6 equiv., the corresponding dibenzylation products were dominant in this reaction. A variety of dibenzylation BODIPYs 43 containing different meso-aryl groups and different methyl arenes at the 3, 5-positions were obtained in 32%–40% yields in the presence 6 equiv. of TBHP and 0.1 equiv. of Cu(OAc)2 at 100℃ (Scheme 16). The direct use of readily available toluene and its derivatives as coupling partners avoids unproductive steps for preactivating the functional group installation, and is therefore attractive. Most of the resulting dyes are highly emissive in the solid state due to the introduction of bulky benzyl groups onto the BODIPY core.

|
Download:
|
| Scheme 16. Regioselective synthesis of 3(, 5)-(di)benzylation of BODIPYs 42 and 43. | |
{kind=link}
4.2. The C–N bond formation
In addition to carbon radicals, similar reactions can occur with heteroatom based free radicals. You and co-workers used AgOAc as an oxidant to oxidize nitrogen-containing compounds such as imidazoles or secondary amine derivatives to nitrogen free radicals, which were then successfully added to BODIPYs at their 3(, 5)-positions [42]. This reaction system was proved to be highly active for the double-amination of a variety of BODIPYs with pyrazole. The double-amination products 46 were synthesized in one-step, using 3, 5-unsubstituted BODIPYs 44 and pyrazole/benzimidazole/arbazole with AgOAc as an oxidant (Scheme 17). Interestingly, the reaction of 44 with aliphatic secondary amines mainly generated mono-amination products in good to almost quantitative yields. The resulting BODIPYs with morpholine groups exhibit specific endoplasmic reticulum (ER)-localization capacities and have negligible cytotoxicities, which would be potential ERtargeting reagents.

|
Download:
|
| Scheme 17. Regioselective synthesis of 46. | |
{kind=link}
4.3. The C–S bond coupling
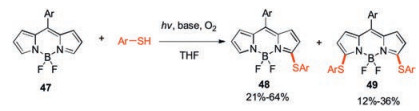
Similar to the halogenated BODIPYs, thiolated BODIPYs have been reported to have good reactivities toward common C-, N- or O-centred nucleophiles through SNAr reactions and aryl boronic acids or aryl-tin reagents through metal catalyzed cross-coupling reactions. Thiols are difficult to undergo oxidative nucleophilic reaction directly with BODIPY. The traditional synthesis of thioether functionalized BODIPYs has been reported by reacting the corresponding halogenated BODIPY with thiols in the presence of a base [24, 25, 43]. Jiao's group found that thiols can easily react with BODIPY under the action of a radical initiator in 2019 [44]. A radical process through hydrogen atom transfer process from thiol to tert-butoxy radical was proposed. By controlling the amount of thiols and initiators, mono-, di- and trithiolation BODIPYs can be obtained (Scheme 18). The monothiolation BODIPY 48 was obtained in best yield when the reaction was carried out in the presence of tert-butylperoxy benzoate (TBPB). DMSO as the solvent was found to be important for the success of this reaction. Interestingly, when the ratio of thiols was increased to 2 equiv., the monothiolation product 48 disappeared and the dithiolation BODIPY 49 was isolated in high yield.

|
Download:
|
| Scheme 18. The synthesis of thiolated BODIPYs 48 and 49. | |
{kind=link}
By increasing the amount of propane-1-thiol to 3 equiv., trithiolated BODIPY 50a was obtained as a major product in a 25% yield. Similarly, trithiolated BODIPY 50b was also synthesized in a 27% yield using the same conditions (Scheme 19). Furthermore, an unsymmetrical BODIPY like the 3-chloroBODIPY 51, which was synthesized using 1.5 equiv. CuCl2 in acetonitrile, was also suitable for this C–H thiolation reaction. The reaction between BODIPY 51 and 1-propanethiol gave only one product 52 in an 80% yield (Scheme 19) and no SNAr substituted product was found.

|
Download:
|
| Scheme 19. The synthesis of thiolated BODIPYs 50 and 52. | |
{kind=link}
At the same time, Xie's group discovered that an autocatalytic reaction can happen on BODIPY with thiophenol under light (Scheme 20), which can also introduce ArS at 3(, 5)-positions of BODIPY [45]. This reaction is also ascribed to proceed through thiol radical process described above. The thioether (pseudohalogen) functionalized BODIPY fluorophores as the key intermediates could easily allow the introduction of a variety of functionalities onto the BODIPY core.

|
Download:
|
| Scheme 20. The photo-induced synthesis of the ArS-substituted BODIPYs. | |
{kind=link}
5. Transition metal catalyzed C–H activation
Apart from outside the two direct C–H functionalization methods above, transition metal catalyzed C–H activation is also an important synthetic method for direct functionalization of the BODIPY core. In 2007, the direct modification of 1, 3, 5, 7-tetramethylBODIPYs was firstly reported by Burgess' group via the palladium-catalyzed oxidative Heck type reaction [46a, 46c]. Afterwards, some other transition metal catalysts based on iridium [46b] or copper [46d, 46e] can also be used to modify the C–H bond of BODIPY directly. However, the selectivities of these metalcatalyzed C–H activation reactions have not been found since the other positions of BODIPY core are blocked by inert groups.
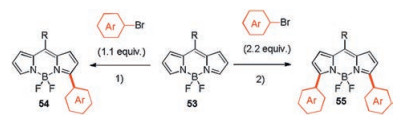
5.1. Regioselective functionalization at the 3(, 5)-positionsIn 2012, Dehaen and co-workers successfully synthesized 3-arylated and 3, 5-diarylated BODIPY dyes in one step (Scheme 21), using palladium-catalyzed C–H arylation of 3, 5-unsubstituted BODIPYs with aryl bromides [47]. This direct, palladium-catalyzed C–H functionalization is a much shorter synthetic protocol and a valuable alternative to the use of halogenated BODIPYs in traditional Suzuki and Stille cross-coupling reactions.

|
Download:
|
| Scheme 21. Direct Pd-catalyzed C–H arylation of meso-substituted BODIPYs 53 with different bromoarenes. R=2, 6-dichlorophenyl, phenyl, or p-nitrophenyl. 1) 5 mol% Pd(OAc)2, 10 mol% HPCy3BF4, 30 mol% pivalic acid (2, 2-dimethylpropanoic acid), 3 equiv. K2CO3, toluene or o-xylene, 110 ℃, 24–48 h. 2) 5 mol% Pd(OAc)2, 10 mol% HPCy3BF4, 30 mol% pivalic acid (2, 2-dimethylpropanoic acid), 3 equiv. K2CO3, toluene or o-xylene, 110 ℃, 4 day. | |
{kind=link}
To illustrate the possibilities of this direct Pd-catalyzed C–H arylation of 3, 5-unsubstituted BODIPYs, unsymmetrical dye 56 and dimeric compound 57 were prepared (Scheme 22). Both compounds were synthesized by performing two sequential C–H arylations, the first with bromobenzene, and the second with 3-bromothiophene or 1, 4-dibromobenzene.

|
Download:
|
| Scheme 22. Synthesis of unsymmetrical BODIPY 56 and a BODIPY dimer 57. | |
{kind=link}
5.2. Regioselective functionalization at the 2(, 6)-positions
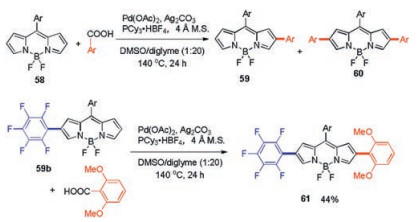
In 2014, You and co-workers successfully synthesized 2-arylated BODIPY 59 and 2, 6-diarylated BODIPY 60 (Scheme 23), using palladium-catalyzed C–H arylation of pyrrolic unsubstituted BODIPYs with benzoic acid in a one-pot reaction [48]. Pd(OAc)2 and PCy3·HBF4 were the superior catalyst/ligand combination. Ag2CO3, and 4 Å molecular sieves (4 Å M.S.) as additives in DMSO/diglyme (1:20, v/v) at 140℃ for 24 h gave the best results.

|
Download:
|
| Scheme 23. Synthesis of 2(, 6)-(di)arylated BODIPYs 59, 60 and unsymmetrical diarylated BODIPY 61. | |
{kind=link}
This decarboxylative C–H arylation allowed the resulting monoarylated BODIPYs to be further arylated by using a different aryl carboxylic acid without preactivation, blockage, protection, and deprotection that are almost inevitable in traditional strategies. Treatment of mono-substituted 59b with 2, 6-dimethoxybenzoic acid afforded an unsymmetrically diarylated BODIPY 61 in a 44% yield. This BODIPY with the D-π-A (donor-π-acceptor) structure has interesting photophysical properties including bathochromic spectra, low band gaps, and large Stokes shifts.
Following this direct C–H arylation, the ortho-nitro group(s) on the 2(, 6)-(di)phenyl substituent(s) of BODIPY 59a and 60a were employed as a useful handle to regioselectively form C–N bond(s) via an intramolecular reductive cyclization, namely the Cadogan reaction (Scheme 24). This method successfully gave indole-fused BODIPYs 62 and 63 for the first time. The indole fused BODIPYs 62 and 63 exhibited remarkably red-shifted absorption and emission spectrum. The maximum peaks of absorption and emission for compound 63 are at 607 nm and 622 nm, respectively. In addition, compound 63 still keeps a large molar extinction coefficient (εmax=145,500 L mol-1 cm-1) and a high quantum yield (Φ=0.71).

|
Download:
|
| Scheme 24. Synthesis of BODIPYs 62 and 63. | |
{kind=link}
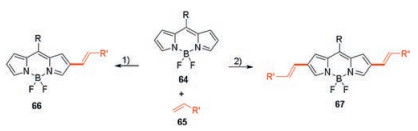
In 2019, Jiao and co-workers successfully synthesized monoalkenylated BODIPYs 66 and dialkenylated BODIPYs 67 using palladium-catalyzed C–H arylation of unsubstituted BODIPYs 64 with styrenes or acrylate 65 (Scheme 25) [49]. Pd(OAc)2 as a catalyst and AgOAc as an oxidant in PivOH at 80℃ for 3 h afforded the β-styrylBODIPY in the best yield.

|
Download:
|
| Scheme 25. Synthesis of monostyrylated BODIPYs 66 and distyrylated BODIPYs 67 through Pd-catalyzed direct C–H styrylation. 1) 0.2 equiv. Pd(OAc)2, 3 equiv. AgOAc, PivOH, 80 ℃. 2) 0.4 equiv. Pd(OAc)2, 5 equiv. AgOAc, PivOH, 80 ℃. | |
{kind=link}
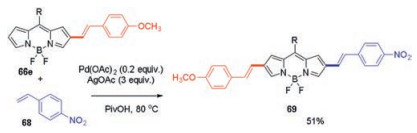
The unsymmetrical distyrylated BODIPY was also synthesized through a further C–H styrylation of the monostyrylated BODIPY (Scheme 26). For example, BODIPY 66e smoothly reacted with 4-nitrostyrene 68 to generate the desired unsymmetrical substituted BODIPY 69 in a 51% yield. Indeed, while the unsubstituted starting BODIPY 64 displays strong green fluorescence, all of the 2-styryl substituted products 66 fluoresce at substantially longer wavelengths in the deep red region, and 2, 6-distyryl substituted BODIPYs 67 and 69 emit in the NIR region (Fig. 4). Increasing the electron-donating ability of the para-substituent in styryl substituted BODIPYs produced bathochromic shifts of both absorption and emission. The bathochromic effects were much more pronounced in fluorescence spectra compared to these of absorption, which caused by a pronounced increase of Stokes shifts of these styryl substituted BODIPYs.

|
Download:
|
| Scheme 26. Synthesis of unsymmetrical distyrylated BODIPYs 69. | |
{kind=link}

|
Download:
|
| Fig. 4. Overlaid normalized absorption (solid lines) and fluorescence emission (dashed lines) spectra of 64 (black), 66e (red) and 69 (blue) in toluene at room temperature. Reproduced from with permission [49a]. Copyright 2019, Wiley Publishing Group. | |
{kind=link}
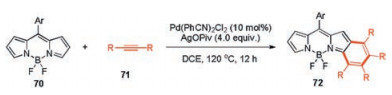
In 2018, You and co-workers successfully synthesized unsymmetrical benzo[b]-fused BODIPYs using a two-fold palladiumcatalyzed C–H arylation of unsubstituted BODIPY 70 with alkynes 71 [50]. Pd(PhCN)2Cl2 as a catalyst and AgOPiv as an oxidant under a N2 atmosphere, afforded the desired product 72 in the best yield (Scheme 27).

|
Download:
|
| Scheme 27. Synthesis of unsymmetrical BODIPY 72. | |
{kind=link}
Among these benzo[b]-fused BODIPYs, multiply aryl substituted derivatives exhibit very low fluorescence quantum yields in dichloromethane. In contrast, a multiply ester-substituted derivative has a moderate fluorescence quantum yield up to 0.24, with the maximum absorption and emission peaks at 590 nm and 614 nm, respectively. Similarly, recently reported phenanthrenefused BODIPYs also showed decreased fluorescence quantum yields with electron rich substituents attached [51].
6. Conclusion and outlookThe facile postfunctionalization of BODIPY dyes, allows one to adequately modulate their physicochemical properties, rendering their applications in highly diverse fields ranging from materials science and biology to medicine. We have summarized several methods for regioselective modification of unsubstituted BODIPYs on the pyrrolic ring through direct C–H bond activations. Developing direct C–H bond transformation methods can avoid complex synthetic routes, reduce the waste of raw materials, and improve the atomic economy of the synthesis. Both the nucleophile substitution reactions and the free radical reactions preferentially react to the α-position of BODIPY to obtain the 3(, 5)-modified derivatives. The transition metal catalyzed method can selectively modify the α- or β-position of BODIPY depending on the conditions. In addition, it is also possible to construct fused rings at β-position of BODIPY by a one-step procedure, and visible light induced radical reaction has been demonstrate as a new powerful method for postfunctionalization of BODIPY dyes by taking advantaging of their excellent photophysical properties. With the rapid development of modern synthetic methods, we can anticipate that more advanced and powerful postfunctionalization methods for BODIPY dyes will rapidly be developed and these latest synthetic methodologies will create more and exciting BODIPY derivatives decorated with various functional groups for various advanced applications.
AcknowledgmentThis work is supported by the National Nature Science Foundation of China (Nos. 21672006, 21672007 and 21871006).
| [1] |
(a) Z. Lei, X. Li, X. Luo, et al., Angew. Chem. Int. Ed. 56 (2017) 2979-2983; (b) J. Zhu, P. Jia, N. Li, et al., Chin. Chem. Lett. 29 (2018) 1445-1450; (c) L. Li, Y. Chen, W. Chen, et al., Chin. Chem. Lett. (2019), doi: http://dx.doi.org/10.1016/j.cclet.2019.04.017; (d) X. Luo, J. Li, J. Zhao, et al., Chin. Chem. Lett. 30 (2019) 839-846; (e) D. Yue, M. Wang, F. Deng, et al., Chin. Chem. Lett. 29 (2018) 648-656. |
| [2] |
(a) A. Loudet, K. Burgess, Chem. Rev. 107 (2007) 4891-4932; (b) G. Ulrich, R. Ziessel, A. Harriman, Angew. Chem. Int. Ed. 47 (2008) 1184-1201. |
| [3] |
(a) H. Lu, J. Mack, T. Nyokong, et al., Coord. Chem. Rev. 318 (2016) 1-15; (b) S. Kolemen, E.U. Akkaya, Coord. Chem. Rev. 354 (2018) 121-134; (c) J. Zhao, K. Xu, W. Yang, et al., Chem. Soc. Rev. 44 (2015) 8904-8939; (d) T. Kowada, H. Maeda, K. Kikuchi, Chem. Soc. Rev. 44 (2015) 4953-4972. |
| [4] |
(a) A. Tursoy, D. Yildiz, E.U. Akkaya, Coord. Chem. Rev. 379 (2019) 47-64; (b) L. Ding, Z. Tian, J. Hou, et al., Chin. Chem. Lett. 30 (2019) 558-562. |
| [5] |
A. Treibs, F.H. Kreuzer, Liebigs Ann. Chem. 718 (1968) 208-223. DOI:10.1002/jlac.19687180119 |
| [6] |
(a) C. Yu, L. Jiao, T. Li, et al., Chem. Commun. 51 (2015) 16852-16855; (b) J. Wang, Q. Wu, S. Wu, et al., Org. Lett. 17 (2015) 5360-5363; (c) C. Yu, Q. Wu, J. Wang, et al., J. Org. Chem. 81 (2016) 3761-3770; (d) N. Chen, W. Zhang, S. Chen, et al., Org. Lett. 19 (2017) 2026-2029. |
| [7] |
I.J. Arroyo, R. Hu, G. Merino, et al., J. Org. Chem. 74 (2009) 5719-5722. DOI:10.1021/jo901014w |
| [8] |
N. Boens, V. Leen, W. Dehaen, Chem. Soc. Rev. 41 (2012) 1130-1172. DOI:10.1039/C1CS15132K |
| [9] |
T. Ueno, Y. Urano, H. Kojima, et al., J. Am. Chem. Soc. 128 (2006) 10640-10641. DOI:10.1021/ja061972v |
| [10] |
Y. Gabe, Y. Urano, K. Kikuchi, et al., J. Am. Chem. Soc. 126 (2004) 3357-3367. DOI:10.1021/ja037944j |
| [11] |
(a) H. Yu, Y. Xiao, L. Jin, J. Am. Chem. Soc. 134 (2012) 17486-17489; (b) C. Yu, Z. Huang, W. Gu, et al., Mater. Chem. Front. 3 (2019) 1823-1832. |
| [12] |
(a) H. Lu, J. Mack, Y. Yang, Z. Shen, Chem. Soc. Rev. 43 (2014) 4778-4832; (b) Y. Ni, J. Wu, Org. Biomol. Chem. 12 (2014) 3774-3791. |
| [13] |
N. Boens, B. Verbelen, W. Dehean, Eur. J. Org. Chem. 2015 (2015) 6577-6595. DOI:10.1002/ejoc.201500682 |
| [14] |
S. Xuan, N. Zhao, X. Ke, et al., J. Org. Chem. 82 (2017) 2545-2557. DOI:10.1021/acs.joc.6b02941 |
| [15] |
(a) W. Sheng, J. Cui, Z. Ruan, et al., J. Org. Chem. 82 (2017) 10341-10349; (b) W. Sheng, Y. Zheng, Q. Wu, et al., Org. Lett. 19 (2017) 2893-2896; (c) W. Sheng, Y. Wu, C. Yu, et al., Org. Lett. 20 (2018) 2620-2623; (d) J. Cui, W. Sheng, Q. Wu, et al., Chem.-Asian J. 12 (2017) 2486-2493; (e) J. Wang, Q. Wu, S. Wang, et al., Org. Lett. 17 (2015) 5360-5363; (f) C. Yu, L. Jiao, T. Li, et al., Chem. Commun. 51 (2015) 16852-16855; (g) Y. Sun, Z. Qu, Z. Zhou, et al., Org. Biomol. Chem. 17 (2019) 3617-3622; (h) Z. Zhou, J. Zhou, L. Gai, et al., Chem. Commun. 53 (2017) 6621-6624. |
| [16] |
(a) Z. Li, E. Mintzer, R. Bittman, J. Org. Chem. 71 (2006) 1718-1721; (b) M. Zhang, E. Hao, Y. Xu, et al., RSC Adv. 2 (2012) 11215-11218. |
| [17] |
R.W. Wagner, J.S. Lindsey, Pure Appl. Chem. 68 (1996) 1373-1380. DOI:10.1351/pac199668071373 |
| [18] |
L. Wu, K. Burgess, Chem. Commun. (2008) 4933-4935. |
| [19] |
L. Jiao, C. Yu, M. Liu, et al., J. Org. Chem. 75 (2010) 6035-6038. DOI:10.1021/jo101164a |
| [20] |
(a) P. Thamyongki, A.D. Bhise, M. Taniguchi, et al., J. Org. Chem. 71 (2006) 903-910; (b) L. Jiao, C. Yu, T. Uppal, et al., Org. Biomol. Chem. 8 (2010) 2517-2519; (c) Z. Wang, C. Cheng, Z. Kang, J. Org. Chem. 84 (2019) 2732-2740. |
| [21] |
C. Yu, L. Jiao, H. Yin, et al., Eur. J. Org. Chem 2011 (2011) 5460-5468. DOI:10.1002/ejoc.201100736 |
| [22] |
V. Lakshmi, M.R. Rao, M. Ravikanth, Org. Biomol. Chem. 13 (2015) 2501-2517. DOI:10.1039/C4OB02293A |
| [23] |
(a) L. Jiao, W. Pang, J. Zhou, et al., J. Org. Chem. 76 (2011) 9988-9996; (b) L. Jiao, J. Li, S. Zhang, et al., New J. Chem. 33 (2009) 1888-1893; (c) X. Zhou, C. Yu, Z. Feng, et al., Org. Lett. 17 (2015) 4632-4635. |
| [24] |
T.V. Goud, A. Tutar, J.F. Biellmann, Tetrahedron 62 (2006) 5084-5091. DOI:10.1016/j.tet.2006.03.036 |
| [25] |
J. Han, O. Gonzalez, A. Aguilar-Aguilar, et al., Org. Biomol. Chem. 7 (2009) 34-36. DOI:10.1039/B818390B |
| [26] |
(a) T. Jiang, P. Zhang, C. Yu, et al., Org. Lett. 16 (2014) 1952-1955; (b) J. Li, Q. Zhang, J. Yin, et al., Org. Lett. 18 (2016) 5696-5699; (c) X. Zhou, Q. Wu, Y. Feng, et al., Chem.-Asian J. 10 (2015) 1979-1986; (d) W. Miao, E. Dai, W. Sheng, et al., Org. Lett. 19 (2017) 6244-6247. |
| [27] |
(a) Z. Feng, L. Jiao, Y. Feng, et al., J. Org. Chem. 81 (2016) 6281-6291; (b) X. Zheng, W. Du, L. Gai, et al., Chem. Commun. 24 (2018) 8834-8837. |
| [28] |
E. Palao, A.R. Agarrabetia, M.J. Ortiz, Org. Lett. 15 (2013) 4454-4457. DOI:10.1021/ol401993p |
| [29] |
(a) L. Jiao, C. Yu, J. Li, et al., J. Org. Chem. 74 (2009) 7525-7528; (b) J. Wang, Y. Wu, W. Sheng, et al., ACS Omega 2 (2017) 2568-2576. |
| [30] |
(a) A. Haefele, C. Zedde, P. Retailleau, et al., Org. Lett. 12 (2010) 1671-1675; (b) M. Zhang, Y. Wu, S. Zhang, et al., Chem. Comm. 48 (2012) 8925-8927. |
| [31] |
V. Leen, V. Zaragozí Gonzalvo, W.M. Deborggraeve, et al., Chem. Commun. 46 (2010) 4908-4910. DOI:10.1039/c0cc00568a |
| [32] |
V. Leen, M. van der Auweraer, N. Boens, W. Dehaen, Org. Lett. 13 (2011) 1470-1473. DOI:10.1021/ol200148u |
| [33] |
M. Zhang, E. Hao, J. Zhou, et al., Org. Biomol. Chem. 10 (2012) 2139-2145. DOI:10.1039/c2ob06689k |
| [34] |
B. Verbelen, S. Boodts, J. Hofkens, et al., Angew. Chem. Int. Ed. 54 (2015) 4612-4616. DOI:10.1002/anie.201410853 |
| [35] |
X. Zhou, Q. Wu, Y. Yu, et al., Org. Lett. 18 (2016) 736-739. DOI:10.1021/acs.orglett.5b03706 |
| [36] |
D. Wang, C. Cheng, Q. Wu, et al., Org. Lett. 21 (2019) 5121-5125. DOI:10.1021/acs.orglett.9b01722 |
| [37] |
B. Verbelen, L. Cunha Dias Rezende, S. Boodts, et al., Chem.-Eur. J. 21 (2015) 12667-12675. DOI:10.1002/chem.201500938 |
| [38] |
B. Tang, F. Lv, K. Chen, et al., Chem. Commun. 55 (2019) 4691-4694. DOI:10.1039/C9CC01602C |
| [39] |
Y. Yu, L. Jiao, J. Wang, et al., Chem. Commun. 53 (2017) 581-584. DOI:10.1039/C6CC08098G |
| [40] |
F. Lv, Y. Yu, E. Hao, et al., Org. Biomol. Chem. 17 (2019) 5121-5128. DOI:10.1039/C9OB00927B |
| [41] |
F. Lv, Y. Yu, E. Hao, et al., Chem. Commun. 54 (2018) 9059-9062. DOI:10.1039/C8CC04679D |
| [42] |
H. Zhang, X. Chen, J. Lan, et al., Chem. Commun. 54 (2018) 3219-3222. DOI:10.1039/C8CC00238J |
| [43] |
V. Leen, P. Yuan, L. Wang, et al., Org. Lett. 14 (2012) 6150-6153. DOI:10.1021/ol3028225 |
| [44] |
F. Lv, B. Tang, E. Hao, et al., Chem. Commun. 55 (2019) 1639-1642. DOI:10.1039/C8CC09821B |
| [45] |
F. Ma, L. Zhou, Q. Liu, et al., Org. Lett. 21 (2019) 733-736. DOI:10.1021/acs.orglett.8b03954 |
| [46] |
(a) C. Thivierge, R. Bandichhor, K. Burgess, Org. Lett. 9 (2007) 2135-2138; (b) J. Chen, M. Mizumura, H. Shinokubo, A. Osuka, Chem.-Eur. J. 15 (2009) 5942-5949; (c) D.K. K ölmel, A. Hcrner, J.A. Castaneda, et al., J. Phys. Chem. C 120 (2016) 4538-4545; (d) W. Ren, H. Xiang, C. Peng, et al., RSC Adv. 8 (2018) 5542-5549; (e) R. Jiang, X. Yang, D. Wu, Org. Biomol. Chem. 15 (2017) 6888-6891. |
| [47] |
B. Verbelen, V. Leen, L. Wang, et al., Chem. Commun. 48 (2012) 9129-9131. DOI:10.1039/c2cc34549h |
| [48] |
L. Luo, D. Wu, W. Li, et al., Org. Lett. 16 (2014) 6080-6083. DOI:10.1021/ol502883x |
| [49] |
(a) J. Wang, Q. Wu, Q. Gong, et al., Adv. Synth. Catal. 361 (2019) 769-777; (b) J. Wang, Y. li, Q. Gong, et al., J. Org. Chem. 84 (2019) 5079-5090. |
| [50] |
X. Yang, L. Jiang, M. Yang, et al., J. Org. Chem. 83 (2018) 9538-9546. DOI:10.1021/acs.joc.8b01239 |
| [51] |
W. Miao, Y. Feng, Q. Wu, et al., J. Org. Chem. 84 (2019) 9693-9704. DOI:10.1021/acs.joc.9b01425 |