 2019, Vol. 30
2019, Vol. 30 


b Institute of Organic Chemistry, University of Mainz, D-55099 Mainz, Germany
Chemosensors to detect and report the organic compounds (analytes) are obtaining an increasing significance in the production processes, the environmental protection and the physiological medicine. In general, chemosensors consist of two major parts: a receptor system and a reporter unit. Moreover, devices for the transformation of the measured signals into electrical currents can be installed.
Supramolecular host-guest complexes are highly suitable to be developed as reporter units. Among well-known macrocyclic hosts, such as crown ethers, calixarenes, veratrylenes and cucurbiturils, the pillararenes appeared as new 'stars' in 2008. Ogoshi et al. published in their pioneering work the first pillar[5]arenes [1]. In 2009 we synthesized the first pillar[6]arenes [2]. Since 2012 pillar[7]arenes [3, 4] and higher pillar[n]arenes (n = 8– 15) are documented [5-7]. A great advantage of pillar[5]arenes and pillar[6]arenes is given by their facile access in high yield and pure condition [8-10]. Their pillar structure contains an interior cavity with a high π electron density suitable for the capturing and the binding of guest molecules. CH…π interactions, hydrophobic and polar interactions inside and at the rim of the cavity are the general driving forces. The encapsulation of the guest molecules is normally proved by NMR (1H chemical shifts, NOE measurements). However, the threading of guest molecules is combined with various alterations of their physical properties. The focus of this article is on the change of the fluorescence intensity by complexation. This includes the ideal situation between a fluorescent and a nonfluorescent state. If necessary, the emitted light can be transformed into electricity. Photocells or charge coupled devices achieve that by applying the photoelectric effect.
The major challenge for the development of chemosensors is to find specific receptor systems and highly sensitive reporter systems for certain analytes. In this report we are giving a survey over selected new results on chemosensors based on pillararene complexes and their fluorescence.
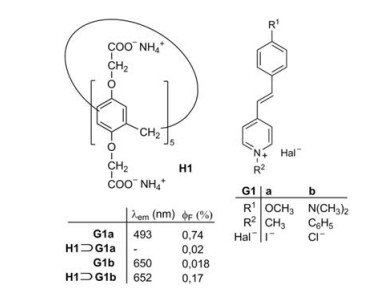
2. Change of the fluorescence intensity by complexationThe fluorescence quantum yield ϕF of a guest or a host molecule can dramatically change by host-guest complexations. The pathway of the fluorescence emission always competes with the radiationless deactivation processes of excited singlet states and the complex formation may favor or disfavor each deactivation route. For example, Fig. 1 shows the complexation of the watersoluble pillar[5]arene H1 and two closely related stilbazolium guest molecules G1a and G1b [11]. Whereas the fluorescence of G1a is almost completely quenched in the complex H1⊃G1a, the fluorescence of G1b is enhanced by a factor close to 10 in the complex H1⊃G1b (Fig. 1). To understand the mechanism for the fluoresence enhancement or quenching, a general introduction on the mechanism of fluorescence is necessary.

|
Download:
|
| Fig. 1. Complexation of the stilbazolium salts G1a and G1b and the pillar[5]arene H1 and the resulting change in fluorescence [11]. | |
{kind=link}
The fluorescence of a supramolecular host guest system normally requires the presence of a fluorophore in the host or in the guest molecule. After the excitation of the molecule (A in the Scheme 1) the excited singlet state S1 of the fluorophore unit can either be radiationless deactivated or emit the fluorescence (F in the Scheme 1) [12]. The internal conversion (IC) directly leads back to the ground state S0, whereas the intersystem crossing (ISC) reaches first the lowest triplet state T1 (Scheme 1). In case of the formation of excimers, where the (S0S1) pairs in a shallow potential energy minimum has no counterpart (S0S0) in the ground state, the deactivation of the excimer to 2 individual S0 is performed by IC or by a fluorescence (F'). In all cases deliberately added quenchers Q can act as new pathway for the deactivation of the excited state.

|
Download:
|
| Scheme 1. Modified Jablonsky diagram for fluorophore units in host-guest systems. | |
{kind=link}
In some cases where the ground state aggregates can be formed, a new fluorescent species might be formed and leads to the aggregation induced emissions (AIE). The absorption A of light leads to an excited singlet state of the aggregate, which can be deactivated by fluorescence (F") or by IC [12].
The quenching process may follow several different routes. Apart from the Förster energy transfer (resonance energy transfer) M* + Q → M + Q*, a polar quenching can occur, in which a photoinduced electron transfer (PET) takes place. The quencher Q can take up an electron in the oxidative quenching M* + Q → M+· + Q-· or it can give off an electron in the reductive quenching M* + Q → M-· + Q+·.
The formation of aggregates quenches the fluorescence in many cases, however, as mentioned above, the opposite effect, AIE in the absence or presence of fluorophore substructures is well established for chemosensors.
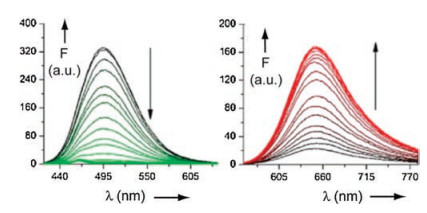
After this general remark on the fluorescence let us come back to the supramolecular system shown in Figs. 1 and 2. The fluorophore units are represented by the stilbazolium ions G1a and G1b [11]. The fluorescence of G1a is almost completely quenched by the threading of G1a into the cavity of H1. A fast electron transfer takes place from the carboxylate groups at the rim of the cavity to the excited singlet states S1 of G1a, with which the deactivation of H1⊃G1a* by fluorescence cannot compete. The excited singlet state S1 of G1b has a much lower dipole moment than G1a [11]. Therefore the electron transfer from the carboxylate groups is less favorable and the major deactivation of the excited singlet state S1 of G1b consists in the twist of the single bonds adjacent to the olefinic double bond of the azastilbene structure. A nonfluorecent twisted intramolecular charge transfer (TICT) state is formed on the way back to the ground state S0. Such a twist is not possible in the cavity of H1⊃G1b. This favors the fluorescence of the complex in comparison to the fluorescence of the free guest molecule G1b.

|
Download:
|
| Fig. 2. Fluorescence titration of G1a (left) and G1b (right) with increasing amounts of H1 in water. Reproduced with permission [11]. Copyright 2016, Elsevier. | |
{kind=link}
The following Sections 3–5 contain host-guest complexations between pillararenes and analytes, which result in alterations of the fluorescence intensities. Free fluorophores and their complexes have often very similar fluorescence band maxima (λem values). However, there are exceptions as discussed below. A formal division between the Sections 3–5 is made according to the origin of the fluorophores.
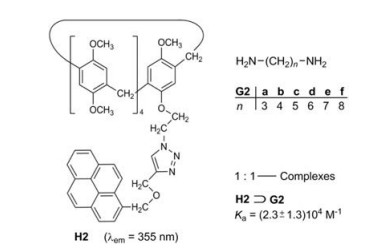
3. Fluorophore substructures in the pillararene hostsStoddart et al. [13] studied the pillararene H2, which bears a pyrene fluorophore on one of its ten alkoxy groups. Primary aliphatic diamines G2a-f can be encapsulated in the cavity of H2 to form the [2]pseudorotaxanes H2⊃G2 (Fig. 3). The association constants Ka of the six 1:1—complexes are in the range of (2.3 ± 1.3) 104 L/mol. The interaction of the excited singlet state S1 of the pyrene moiety and the encapsulated diamines causes a significant fluorescence quenching. Such a quenching effect of amines is wellknown and can be explained by an electron transfer from the amine to the excited arene [14]. Primary monoamines also thread into the cavity of H2, but their quenching effect is smaller. The titration of amines with pyrene employing the same conditions did not result in a fluorescence quenching [13]. The encapsulation is obviously a precondition for a close interaction of pyrene and amine.

|
Download:
|
| Fig. 3. Complexation of the diamines G2a-G2f and the pillar[5]arene H2. | |
{kind=link}
The sensitivity and selectivity of the host H2 towards the diamines G2 as guests is moderate, but H2/G2 represented the first chemosensor on a pillararene basis and had a very stimulating impact on this research area.
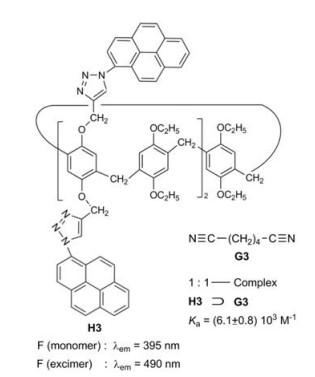
Ogoshi et al. investigated pillar[5]arenes bearing 2, 4 or 10 pyrene fluorophores fixed on the alkoxy chains [15]. In addition to the monomer fluorescence the compounds exhibit a strong excimer fluorescence, whose intensity is enhanced, when adiponitrile G3 as guest is threaded into the cavity of H3 (Fig. 4). The ratio of the intensity of the excimer fluorescence to the intensity of the monomer fluorescence is 2.9:1 for the pure host H3. In the course of the titration with adiponitrile this ratio is enlarged to 3.5:1. The complexation rigidifies the pillar structure and limits molecular vibrations responsible for IC, thus leading to the enhancement of the fluorescence intensity.

|
Download:
|
| Fig. 4. Complexation of adiponitrile G3 and pillar[5]arene H3. | |
{kind=link}
Two quinoxaline building blocks were fixed in the host H4 over thiophene rings to a benzene ring by us [16], which is a part of a pillar[5]arene macrocycle. Terminal dinitriles G3 and G4a–d form 1:1-complexes with H4 (Fig. 5). The energy-lowest absorptions of these complexes are charge transfer (CT) bands with an intramolecular electron transfer from the methoxy substituted benzene rings to the quinoxaline subunits. The fluorescence corresponds to the reverse process. In comparison to H4 the fluorescence of H4⊃G4a is reduced, but the emission of H4⊃G4b and the other dinitrile complexes is enhanced (Fig. 6) [16].

|
Download:
|
| Fig. 5. Complexation of the dinitriles G3 and G4a–d in the pillar[5]arene H4. | |
{kind=link}

|
Download:
|
| Fig. 6. Fluorescence titration of H4 with different dinitriles in CDCl3. (a) Decrease of intensity with increasing amounts of G4a; inset: Binding isotherm of H4 with G4a. (b) Increase of intensity with increasing amounts of G4b; inset: binding isotherm of H4 with G4b. Reproduced with permission [16]. Copyright 2018, American Chemical Society. | |
{kind=link}
An explanation of this unexpected behavior is given by the structures of the complexes and their electrostatic potential maps. TDDFT calculations predict a deep encapsulation of the cyano groups of malonitrile (G4a) in the cavity of H4, whereas the two cyano groups of H4⊃G4b are located at the rim of the cavity. A crystal structure of H4⊃G4b is depicted in Fig. 7.

|
Download:
|
| Fig. 7. Crystal structure of the complex H4 ⊃ G4b. (B: benzene rings of H4, Q: quinoxaline units, T: thiophene rings, the dashed lines indicate the interactions between the CN and the OCH3 groups). Reproduced with permission [16]. Copyright 2018, American Chemical Society. | |
{kind=link}
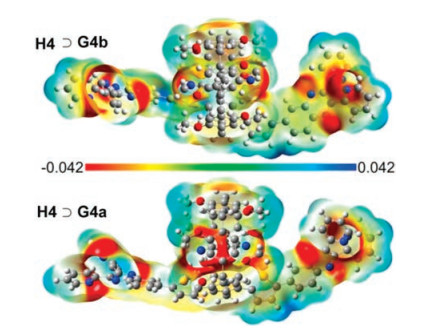
Electron-withdrawing groups such as CN induce a negative electrostatic potential in their region. Accordingly, the return of an electron from the quinoxaline substructure in this area is impeded. The effect is called polarity induced fluorescence quenching. On the contrary cyano groups at the rim of the cavity induce a positive electrostatic potential in the interior cavity and enhance the fluorescence quantum yield (Fig. 8).

|
Download:
|
| Fig. 8. Electrostatic potentials: electron isodensity surfaces of H4⊃G4b (top) and H4⊃G4b (bottom). Copied with permission [16]. Copyright 2018, American Chemical Society. | |
{kind=link}
Zheng et al. found an interesting method to identify traces of TNT in the air [17]. The pillar[6]arene derivatives H5a–c exhibit AIE in the solid state (λem = 475 nm). The fluorescence of a suspension of H5a in H2O/THF (95:5) has a quantum yield ϕF of 24%. Electrondeficient aromatics become included in the electron-rich cavity of H5a–c and cause a strong fluorescence quenching. TNT (G5a) in H5a shows a quenching rate of 60%. Other nitroaromatics have a smaller effect, 2, 4, 6-trinitrophenol (picric acid G5b) for example 33%. The high sensitivity for TNT promises an interesting security technique to detect TNT in the air [17] (Fig. 9).

|
Download:
|
| Fig. 9. Pillar[6]arenes H5a–c bearing included TNT (G5a) or picric acid (G5b). | |
{kind=link}
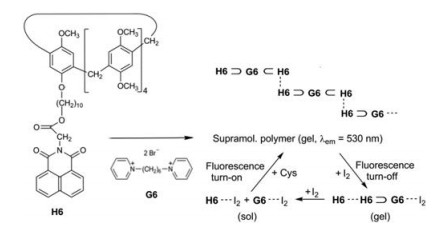
Zhang et al. studied the supramolecular polymer obtained from pillar[5]arene H6 and the bispyridinium salt G6 [18]. The guest molecules G6 are encapsulated on both ends in host cavities. Moreover π…π interactions between the naphthalimide units establish the generation of supramolecular chains. Hot solutions (T > Tgel) in DMSO/H2O (9:1) exhibit a negligible fluorescence. However, if by lowering the temperature, the stable gel state is reached, an intense yellow AIE fluorescence (λem = 530 nm) can be observed. Addition of iodine (< 1 equiv.) maintains the gel state but quenches the fluorescence by charge-transfer interactions G6…I2. Higher amounts of iodine (I2 > 1 equiv.) effect an additional CT interaction between the naphthalimide substructures and I2 and trigger a disassembly. When then the iodine is consumed by the oxidation of added L-cysteine, the gel state and its fluorescence is restored (Scheme 2) [18].

|
Download:
|
| Scheme 2. Self-assembly of H6 and G6 to a supramolecular polymer, which fluoresces in the gel state, shows a fluorescence turn-off in the presence of small amounts of iodine and a new turn-on of the emission, when the iodine is consumed by added L-cysteine. | |
{kind=link}
Yang et al. prepared a conjugated macrocyclic polymer H7 (Fig. 10) by a Sonogashira coupling of a functionalized pillar[5] arene and an ethane bearing four 4-ethynylphenyl substituents [19]. The obtained insoluble, dark green powder has a reticular structure and exhibits a two-photon fluorescence at about 537 nm (ϕF = 25.6%). UV excitation (300–400 nm) leads to a virtual energy level and excitation at the edge of the NIR region (650–800 nm) generates then the excited state which gives an AIE. Apart from various ions, organic molecules can provoke an efficient quenching of the emission. In particular 4-aminoazobenzene (G7), which is a dangerous carcinogenic, yellow dye, gives a quenching rate of 98.5%, which renders H7 to an attractive two-photon fluorescent sensor.

|
Download:
|
| Fig. 10. Reticular polymer H7 containing pillar[5]arene building blocks, that can complex 4-aminoazobenzene (G7). | |
{kind=link}
Polymers which contain many pillararene units can be excellent receptor systems, but when their fluorescence intensity is not considerably altered by the complexation of guest molecules, they are inadequate reporter systems for fluorescent chemosensors [20].
4. Fluorescent guest moleculesThe water-soluble pillar[5]arene H8 represents an excellent fluorescent chemosensor for dopamine hydrochloride (G8) (Fig. 11) [21]. The fluorescence intensity of G8 decreases with increasing concentration of H8. The detection limit for G8 is very low at 0.03 μg/mL.

|
Download:
|
| Fig. 11. Complexation of dopamine hydrochloride (G8) and the water-soluble pillar[5]arene H8. | |
{kind=link}
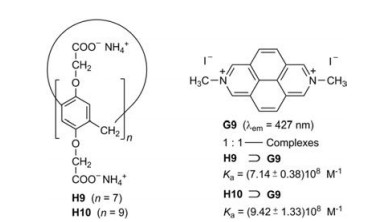
Due to their wide cavities the water-soluble pillar[7]- and pillar [9]arenes H9 [22] and H10 [23] form very stable inclusion complexes with the large diazapyrenium salt G9 (Fig. 12). The fluorescence of G9 decreases continuously on titration with H9 or H10 in water. The obtained association constants Ka are almost 109 L/mol. The assembly and disassembly of the 1:1-complex H10⊃G9 can be reversibly controlled by sequential addition of HCl and NaOH [23].

|
Download:
|
| Fig. 12. Complexation of the diazapyrenium salt G9 and the pillar[n]arenes H9 (n = 7) and H10 (n = 9), respectively. | |
{kind=link}
The size of the macrocyclic hosts has a strong influence on the association constants Ka. Fig. 13 summarizes the Ka values of the inclusion complexes of the pillar[n]arenes H1 and H9-H12 of different size n of the macrocycles and paraquat (dimethylviologen, G10) as guest [24-28]. Paraquat is a toxic herbizide, whose detection in water is important. The highest Ka value is obtained for n = 7. Further increase of the size of the macrocycles weakens the association strength of the 1:1-complexes. A [3] pseudorotaxane can be established for n = 10. It consists of H12 and two guest molecules G10. Accordingly, two Ka values can be obtained (Fig. 13).

|
Download:
|
| Fig. 13. Binding constants of water-soluble pillar[n]arene complexes with paraquat (G10): H1⊃G10, H11⊃G10, H9⊃G10, H10⊃G10 and H12⊃(G10)2 and their dependence on the size of the macrocyclic rings. | |
{kind=link}
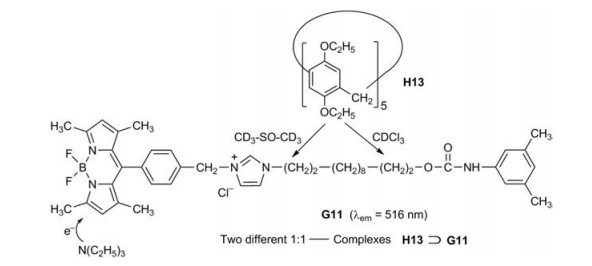
Pillar[5]arene H13 and the boron dipyrromethane dye G11 as fluorophore form a [2]rotaxane [29]. The "wheel" H13 moves over the dumb-bell-shaped alkylene axle of G11 under multiple external stimuli. According to 1H NMR measurements it is in pure CDCl3 on the urethane (carbamate) side and in pure CD3-SO-CD3 on the imidazolium side. The fluorescence decreases by moving from the imidazolium side to the urethane side, because the nonradiative internal conversion is in polar solvents less effective. Increasing amounts of trimethylamine decrease the fluorescence of H13⊃G11 further (Figs. 14). Neutralization of the amine by trifluoroacetic acid results in a complete recovery of the original fluorescence intensity. A PET from the amine to the excited boron dipyrromethane unit may be responsible for this effect [30, 31].

|
Download:
|
| Fig. 14. Combination of a fluorescent sensor and a molecular shuttle. | |
{kind=link}
Huang et al. studied the complexation of a rod-coil guest molecule G12 and the pillar[5]arene H1 [32]. The 2:1-complexe (H1)2⊃G12 represents a supraamphiphile with the qinquephenyl chain as hydrophobic part and the alkoxy chains with terminal imidazolium rings as hydrophilic part (Figs. 15). G12 forms in water sheet-like aggregates of about 1 × 0.1 μm2, whereas the complex (H1)2⊃G12 generates vesicles with a diameter of about 0.2 μm. The fluorescence of the complex in water is enhanced by a factor of about 20 in comparison to the guest in the absence of host. The pillar[5]arene macrocycles weaken the π…π stacking of the quinquephenyl partial structure.

|
Download:
|
| Fig. 15. Complexation of G12 and model compound G13 with host H1 as multi stimuli sensor. | |
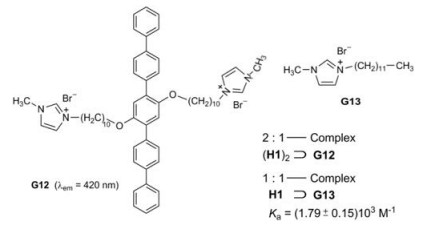{kind=link}
When paraquat G10 is added to (H1)2⊃G12, the fluorescence decreases significantly, because the binding constant of H1⊃G10 is more than 40 times higher than that of complex H1⊃G13. The latter is used as likely Ka value for the complexation of H1 and G 12.
The supraamphiphile (H1)2⊃G12 can also be used as a fluorescent sensor for low pH values. Addition of H+ protonates the carboxylate anions of H1, the host precipitates in the aqueous solution and the fluorescence decreases [32].
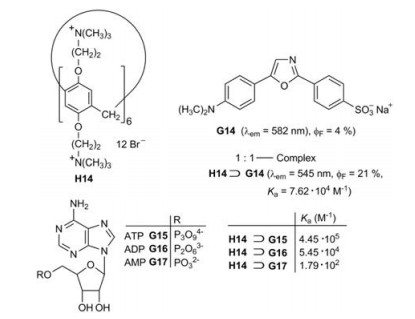
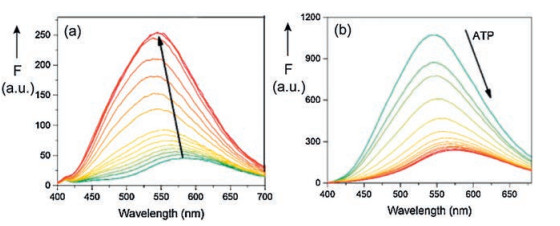
A sensor for ATP was developed on the basis of pillar[6]arene H14 as host and the dye dapoxyl sodium sulfonate (G14) as guest (Fig. 16) [33]. The 1:1-complex H14⊃G14 exhibits an enhanced fluorescence in comparison to the free fluorophore G14 in buffered aqueous solution. Moreover, the fluorescence band of the complex is considerably shifted to lower wavelength (Fig. 17a). According to molecule calculations (MOPAC 2016) the guest G14 is attached with its electron-rich sulfonate function to the electron-deficient ammonium functions at the rim of the cavity of the host H1.

|
Download:
|
| Fig. 16. Development of a sensor for ATP on the basis of a fluorescence indicator displacement. | |
{kind=link}

|
Download:
|
| Fig. 17. (a) Variation of the fluorescence spectrum of dapoxyl sodium sulfonate (14) by addition of pillar[6]arene H14 in HEPES buffer. (b) Variation of the fluorescence spectrum of H14⊃G14 by additional ATP [33]. Copied with permission [33]. Copyright 2017, Elsevier. | |
{kind=link}
The complex H14⊃G14 is suitable for the fluorescence indicator displacement (FID) upon the addition of adenosine-5'-triphosphate (ATP, G15). The turn-off fluorescence spectrum is depicted in Fig. 17b. The selectivity as ATP sensor is demonstrated by the comparison to the complexes with ADP (G16) and AMP (G17), which have much lower binding constants Ka [33].
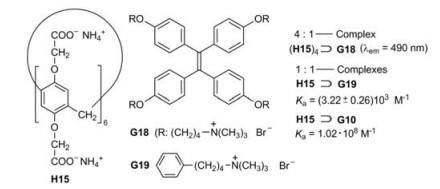
The water-soluble pillar[6]arene H15 (i.e., H11) forms a fourfold [2]pseudorotaxane by complexation with the tetraphenylethene derivative G18 [34]. The related mono[2]pseudorotaxane H15⊃G19 has an aggregation constant of about 3000 L/mol, which represents a reliable value for (H15)4⊃G18 as well. The molecules G18 are almost non-emissive in the excited singlet state S1 in water. They lose their energy by IC, which is triggered by rotations of the benzene rings. However, these rotations are hampered in the complex (H15)4⊃G18. Therefore the titration of G18 with H15 leads to a gradual increase of the fluorescence (Fig. 18). When paraquat (G10) is added, G18 is displaced from the complex (H15)4⊃G18 owing to its much higher Ka value of H15⊃G10 [34].

|
Download:
|
| Fig. 18. Complexation of the guests G18, G19 and G10 in the water-soluble pillar[6]arene H15 as host. | |
{kind=link}
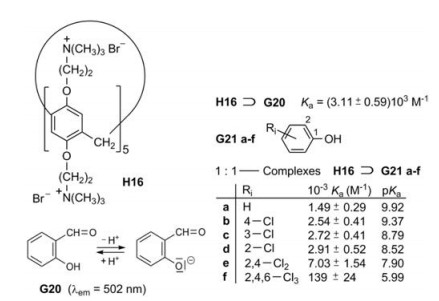
Huang et al. applied salicylaldehyde (G20) respectively its anion as fluorescence indicator [35]. The host H16 assists the deprotonation of the OH group; that means salicylaldehyde becomes a stronger acid by complexation, because its anion binds stronger to the pillar[5]arene H16 than the electroneutral molecule. The polar interaction of O- and (CH3)3N+ provides an easy explanation. Salicylaldehyde itself shows a weak fluorescence in a phosphate buffered solution at pH 7.2, but its anion fluoresces strongly under these conditions. When the phenols G21a-f are added to H16⊃G21, the fluorescence decreases in the same order as the pKa values of the phenols decrease, because the association constants increase in this order [35]. Electron-withdrawing substituents lower the pKa value, so that more phenolate anions are present in the dissociation equilibrium. Accordingly the polar interactions are enhanced in the cavity and the fluorescence of the complexes H16⊃G21a-f is decreased (Fig. 19).

|
Download:
|
| Fig. 19. Complexation of salicylaldehyde (G20) and H16 and related complexations of phenol (G21a) and chlorinated phenols (G21b–f). | |
{kind=link}
Bitter et al. studied the possibility of the recognition of amino acids by pillararene-based FID assays [36]. H1 served as host and the fluorescent naphthalimides G22a-G22c as guests (Fig. 20). The fluorescence of the 1:1—complexes H1⊃G22 is quenched in buffered aqueous solution, because PET is very efficient from the ten negative charges at the rim of the cavity to the excited guest molecules. Cadaverine (G23), lysine (G24) or arginine (G25) can displace the naphthalimides G22 and provoke a turn-on of the fluorescence. The percentage of the fluorescence regeneration is determined by the proportion of the displaced indicator (Fig. 20) [36].

|
Download:
|
| Fig. 20. Complexation of the naphthalimides G22a–c and the water-soluble H1 and FID by cadaverine (G23), lysine (G24) or arginine (G25). | |
{kind=link}
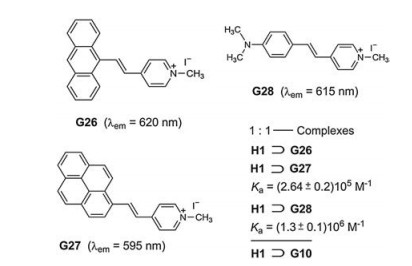
The high tendency of basic amino acids to thread into the cavity of H1 was originally observed by Li et al. [37]. Bitter et al. studied also the complexation of H1 and the stilbazolium dyes G26-G28. The most interesting result was found for H1⊃G28, which shows a 28-fold enhancement of the fluorescence intensity in comparison to the free stilbazolium iodide G28 [38]. The emission of G28 is allocated to the locally excited, planar singlet state, whereas the intramolecular charge transfer (ICT) and TICT state of G28 are nonemissive in water (Fig. 21).

|
Download:
|
| Fig. 21. Complexation of the stilbazolium dyes G26-G28 and H1. FID assay based on H1⊃G28 for the detection of paraquat (G10). | |
{kind=link}
The complex H1⊃G28 proved to be suitable for FID by addition of paraquat (G10). Dethreading of G28 initiates almost complete turn-off fluorescence. The quenching results from an efficient PET.
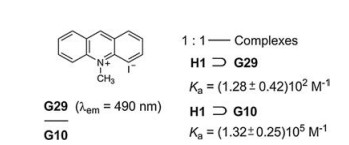
Another recognition and sensing of paraquat (G10) was performed with H1 and 10-methylacridinium iodide (G29) [39]. PET causes a fluorescence quenching in the complex H1⊃G29. Since the association constant Ka of H1⊃G10 is more than 103 times larger than Ka of H1⊃G29, the dethreading of G29 and its fluorescence emission is guaranted. While the sensing of paraquat is based on the turn-off fluorescence in Fig. 21, it is based on the contrary on turn-on fluorescence here (Fig. 22).

|
Download:
|
| Fig. 22. Complexation of 10-methylacridinium iodide (G29) and H1 and FID assay for the sensing of paraquat (G10). | |
{kind=link}
Li et al. used safranine T (G30) as an electron-deficient dye indicator for their FID assay and grafted the pillar[5]arene H17 with its terminal hydrazide group onto graphene as a singlet quencher (Fig. 23) [40]. The fluorescence intensity of G30 is reduced by the energy transfer G30* → Graphene to 5%. The conjugate system Graphene—H17 can be utilized as the highly effective fluorescence recognition platform for paraquat (G10) both in vitro and in vivo. The other applied dipyridyl compounds G31-G34 do not give comparable turn-on fluorescence [40].

|
Download:
|
| Fig. 23. Complexation of safranine T (G30) and H17 grafted onto graphene. FID of G30 by paraquat (G10) and the related dipyridyl compounds G31-G34. | |
{kind=link}
A pillar[5]arene functionalized reduced graphene oxidewas used for a sensitive and selective fluorescent chemosensor for acetaminophen(G35), an analgetic and antipyretic drug, which is also known under the name paracetamol (Fig. 24) [41]. H18 complexes the dye acridine orange (G35), whose fluorescence is quenched. Displacement of the dye by acetaminophen/paracetamol stops the quenching and the bright fluorescence of acridine orange (G35) returns (Fig. 24).

|
Download:
|
| Fig. 24. Complexation of acridine orange (G35) and H18 grafted onto reduced graphene oxide. FID assay for the proof of acetaminophen/paracetamol (G36). | |
{kind=link}
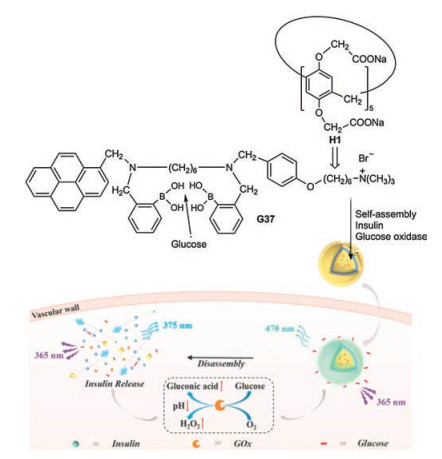
Recently an integrated glucose sensing and insulin delivery system was developed based on the water-soluble H1 (Scheme 3) [42]. The guest G37 of the complex H1⊃G37 contains a twofold phenylboronic acid partial structure, which represents a highly selected receptor of glucose. G37 threads with its alkylammonium chain into the host. H1⊃G37—glucose forms vesicles, in which the π-π stacking of the pyrene moieties results in an excimer fluorescence at about 470 nm. The selective recognition of glucose causes the disruption of the vesicles combined with the disaggregation of the pyrene stacks. Consequently the excimer emission is reduced and the monomer emission (~375 nm) enhanced at the hyperglymic level. When the vesicles are loaded with insulin and glucose oxidase, the disruption of the vescicular structure can be coupled with the release of insulin triggered by a sufficiently high glucose concentration. A low level pH and a high H2O2 environment, which generated during the enzyme-promoted oxidation of glucose to gluconic acid, fascilitates the disassembly of the vesicles. The whole system represents a nanoplatform, which works as glucose sensor and controlled insulin delivery actuator.

|
Download:
|
| Scheme 3. Complexation of H1 and guest G37, which is a selective receptor of glucose. Nanoplatform which works as glucose sensor and insulin delivery actuator. Reproduced with permission [42]. Copyright 2018, John Wiley and Sons. | |
{kind=link}
5. Systems without additional fluorophores
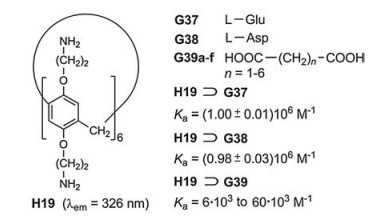
In rare cases, the incorporation of special fluorophores can be dispensed with. The water-soluble pillar[6]arene H19, which bears twelve amino groups, generates highly stable inclusion complexes with the acidic amino acids glutamine (G37) and asparagine (G38) (Fig. 25) [43]. Among the twenty natural, proteinogenic amino acids this property is highly selective. The carboxylate ions of G37 and G38 interact with the protonated portals of the host cavities. This effect represents the dominant host-guest interaction at pH ~6.0 in water. The dicarboxylic acids G39a–f form complexes of much lower binding constants Ka. Titration of H19 with increasing amounts of G37 or G38 leads to a minor lowering of the fluorescence intensity. In contrast to the high selectivity of the sensing, the sensitivity is low.

|
Download:
|
| Fig. 25. Complexation of the amino dicarboxylic acids G37 and G38 in the cavity of H19 and the comparably much weaker complexations of the dicarboxylic acids G39a–f. | |
{kind=link}
6. Conclusions and outlook
Pillar[n]arenes represent novel, highly promising receptor systems (hosts H) for applications in fluorescent chemosensors for organic compounds (guests G). The analytes tobe detected have to thread into the cavity of the pillar[n]arenes and generate stable complexes H⊃G. The fluorescence as reporter method requires a turn-on or turn-off mode – at least a considerable change of the fluorescence intensity (fluorescence quantum yield) in the complexation. Apart from the normal emission at the electron transition S1 → S0 of a fluorophore, excimer fluorescence, twophoton fluorescence and AIE can be used. The fluorophore can be provided by the host or by the guest. The necessary alteration of the fluorescence intensity can have different reasons:
(ⅰ) The radiationless deactivation of the excited singlet state S1 by vibrations or torsions is restricted for encapsulated guests compared to free guest molecules, which means the fluorescence is amplified.
(ⅱ) The electrostatic potentials at the surface of the guest change by the encapsulation, so that polar interactions between guest and host can become important in the interior cavity and at its portals. Depending on the electron distribution, this can enhance or reduce the fluorescence, because PET is a major competition route to the fluorescence.
(ⅲ) Aggregation induced quenching of the fluorescence and AIE.
A well-established procedure is given by FID. A fluorescent dye becomes encapsulated and loses its emission till a nonfluorescent analyte displaces the dye in the complex and the recurrence of the fluorescence indicates the analyte. An important condition for this method is, that the binding constant (association constant) Ka of the analyte is larger than that of the dye.
Onthewhole, the sensing of organic compounds by complexation in pillar[n]arenes is much less studied than the sensing of ions [44, 45]. Nevertheless, there are highly attractive examples for the tracking of organic compounds by pillar[n]arene sensors: dopamine, ATP, acetaminophen(paracetamol), glucosecarriers, TNT, etc.
Important objectives for future technical applications of pillar [n]arenes in fluorescent chemosensors is to scrutinize the selectivity and sensitivity of the pillararene-analyte complexes and to extend this strategy to covalently bound pillararene-analyte systems [46].
AcknowledgmentsWe are grateful to the National Natural Science Foundation of China (Nos. 21572069, 21772045), the National Key Research and Development Program of China (No. 2016YFA0602900), and the Natural Science Foundation of Guangdong Province, China (No. 2018B030311008).
| [1] |
T. Ogoshi, S. Kanai, S. Fujinami, et al., J. Am. Chem. Soc. 130 (2008) 5022-5023. DOI:10.1021/ja711260m |
| [2] |
D. Cao, Y. Kou, J. Liang, et al., Angew. Chem. 121 (2009) 9901-9903 Angew. Chem. Int. Ed. 48 (2009) 9721-9723.
|
| [3] |
Y. Chen, H.Q. Tao, Y.H. Kou, et al., Chin. Chem. Lett. 23 (2012) 509-511. DOI:10.1016/j.cclet.2012.03.026 |
| [4] |
C. Han, Z. Zhang, X. Chi, et al., Acta Chim. Sin. 70 (2012) 1775-1778. DOI:10.6023/A12060296 |
| [5] |
X.B. Hu, Z. Chen, L. Chen, et al., Chem. Commun. 48 (2012) 10999-11001. DOI:10.1039/c2cc36027f |
| [6] |
S.T. Schneebeli, C. Cheng, K.J. Hartlieb, et al., Chem. -Eur. J. 19 (2013) 3860-3868. DOI:10.1002/chem.201204097 |
| [7] |
T. Ogoshi, N. Ueshima, F. Sakakibara, et al., Org. Lett. 16 (2014) 2896-2899. DOI:10.1021/ol501039u |
| [8] |
(a) D. Cao, H. Meier, Asian J. Org. Chem. 3 (2014) 244-262; (b) T. Xiao, W. Zhong, L. Zhou, et al., Chin. Chem. Lett. 30 (2019) 31-36. |
| [9] |
(a) M. Wang, X. Du, H. Tian, et al., Chin. Chem. Lett. 30 (2019) 345-348; (b) D. Cao, H. Meier, Synthesis 47 (2015) 1041-1056. |
| [10] |
(a) T. Ogoshi, T.-A. Yamagishi, Y. Nakamoto, Chem. Rev.116 (2016) 7937-8002; (b) X.Y. Lou, Y.W. Yang, Adv. Opt. Mater. 6 (2018) 18006686; (c) M.X. Wu, Y.W. Yang, Polym. Chem. 10 (2019) 2980-2985; (d) X.Y. Jin, N. Song, X. Wang, et al., Sci. Rep. 8 (2018) 4035. |
| [11] |
M. Bojtár, Z. Szakács, D. Hessz, et al., Dyes Pigm. 133 (2016) 415-423. DOI:10.1016/j.dyepig.2016.06.030 |
| [12] |
N.J. Turro, V. Ramamurthy, J.C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, USA, 2010.
|
| [13] |
N.L. Strutt, R.S. Forgan, J.M. Spruell, et al., J. Am. Chem. Soc. 133 (2011) 5668-5671. DOI:10.1021/ja111418j |
| [14] |
V. Kawade, A. Kumbhar, A. Sapre, A. Kumbhar, Chem. Phys. Lett. 583 (2013) 198-202. DOI:10.1016/j.cplett.2013.07.071 |
| [15] |
T. Ogoshi, D. Yamafuji, D. Kotera, et al., J. Org. Chem. 77 (2012) 11146-11152. DOI:10.1021/jo302283n |
| [16] |
W. Cui, L. Wang, L. Xu, et al., J. Phys. Chem. Lett. 9 (2018) 1047-1052. DOI:10.1021/acs.jpclett.8b00037 |
| [17] |
J.H. Wang, H.T. Feng, Y.S. Zheng, Chem. Commun. 50 (2014) 11407-11410. DOI:10.1039/C4CC05189K |
| [18] |
Q. Lin, K.P. Zhong, J.H. Zhu, et al., Macromolecules 50 (2017) 7863-7871. DOI:10.1021/acs.macromol.7b01835 |
| [19] |
X. Li, Z. Li, Y.W. Yang, Adv. Mater. 30 (2018) 1800177. DOI:10.1002/adma.201800177 |
| [20] |
W. Cui, H. Tang, L. Xu, et al., Macromol. Rapid Commun. 38 (2017) 1700161. DOI:10.1002/marc.201700161 |
| [21] |
X.D. Xiao, L. Shi, L.H. Guo, et al., Spectrochim. Acta A 173 (2017) 6-12. DOI:10.1016/j.saa.2016.08.050 |
| [22] |
Z. Li, J. Jang, F. Huang, Chin. J. Chem. 36 (2018) 59-62. DOI:10.1002/cjoc.201700601 |
| [23] |
Z. Li, G. Yu, J. Jang, Org. Chem. Front. 4 (2017) 115-118. DOI:10.1039/C6QO00579A |
| [24] |
T. Ogoshi, M. Hashizume, T.A. Yamagishi, et al., Chem. Commun. 46 (2010) 3708-3710. DOI:10.1039/c0cc00348d |
| [25] |
G. Yu, X. Zhou, Z. Zhang, et al., J. Am. Chem. Soc. 134 (2012) 19489-19497. DOI:10.1021/ja3099905 |
| [26] |
Z. Li, J. Jang, G. Yu, et al., Org. Lett. 16 (2014) 2066-2069. DOI:10.1021/ol500686r |
| [27] |
Z. Li, J. Jang, G. Yu, et al., Chem. Commun. 50 (2014) 2841-2843. DOI:10.1039/c3cc49535c |
| [28] |
X. Chi, G. Yu, L. Shao, et al., J. Am. Chem. Soc. 138 (2016) 3168-3174. DOI:10.1021/jacs.5b13173 |
| [29] |
N. Sun, X. Xiao, W. Li, et al., Adv. Sci. 2 (2015) 1500082. DOI:10.1002/advs.201500082 |
| [30] |
M.N. Padden-Row, Acc. Chem. Res. 27 (1994) 18-25. DOI:10.1021/ar00037a003 |
| [31] |
A.P. De Silva, H.Q.N. Gunaratne, C.P. McCoy, Chem. Commun. 21 (1996) 2399-2400. |
| [32] |
Y. Yao, X. Chi, Y. Zhou, et al., Chem. Sci. 5 (2014) 2778-2782. DOI:10.1039/c4sc00585f |
| [33] |
M. Bojtár, J. Kozma, Z. Szakács, et al., Sens. Actuators B 248 (2017) 305-310. DOI:10.1016/j.snb.2017.03.163 |
| [34] |
P. Wang, X. Yan, F. Huang, Chem. Commun. 50 (2014) 5017-5019. DOI:10.1039/c4cc01560f |
| [35] |
B. Hua, L. Shao, G. Yu, et al., Chem. Commun. 52 (2016) 10016-10019. DOI:10.1039/C6CC04919B |
| [36] |
M. Bojtár, A. Paudies, D. Hessz, et al., RSC Adv. 6 (2016) 86269-86275. DOI:10.1039/C6RA15003A |
| [37] |
C. Li, J. Ma, L. Zhao, et al., Chem. Commun. 49 (2013) 1924-1926. DOI:10.1039/c3cc38622h |
| [38] |
M. Bojtár, Z. Szakács, D. Hessz, et al., RSC Adv. 5 (2015) 26504-26508. DOI:10.1039/C4RA14809F |
| [39] |
P. Wang, Y. Yao, M. Xue, Chem. Commun. 50 (2014) 5064-5067. DOI:10.1039/C4CC01403K |
| [40] |
X. Mao, T. Liu, J. Bi, et al., Chem. Commun. 52 (2016) 4385-4388. DOI:10.1039/C6CC00949B |
| [41] |
G. Zhao, L. Yang, S. Wu, et al., Biosens. Bioelectron. 91 (2017) 863-869. DOI:10.1016/j.bios.2017.01.053 |
| [42] |
M. Zuo, W. Qian, Z. Xu, et al., Small 14 (2018) 1801942. DOI:10.1002/smll.201801942 |
| [43] |
Q. Duan, W. Zhao, K. Lu, Tetrahedron Lett. 58 (2017) 4403-4406. DOI:10.1016/j.tetlet.2017.10.025 |
| [44] |
T.L. Mako, J.M. Racicot, M. Levine, Chem. Rev. 119 (2019) 322-477. DOI:10.1021/acs.chemrev.8b00260 |
| [45] |
J.F. Chen, Q. Lin, Y.M. Zhang, et al., Chem. Commun. 53 (2017) 13296-13311. DOI:10.1039/C7CC08365C |
| [46] |
Q. Lin, Y.Q. Fan, G.F. Gong, et al., ACS Sustain. Chem. Eng. 6 (2018) 8775-8781. DOI:10.1021/acssuschemeng.8b01124 |