 2019, Vol. 30
2019, Vol. 30 


b Shaanxi Institute of Flexible Electronics(SIFE), Northwestern Polytechnical University(NPU), Xi'an 710072, China
Enzymes play a vital role in living system due to their highly efficient catalytical activities that accelerate the biochemical reactions [1]. Abnormal enzyme activity could lead to numerous diseases such as cancers, Parkinson's disease (PD) and Alzheimer's disease (AD) [2]. However, the most commonly used traditional methods for detecting the enzymatic reactions are not able to monitor the corresponding activity in real time [3]. Therefore, it is necessary to design and develop a tool that can directly detect enzyme activity in vivo, more importantly, in a non-invasive and real-time manner.
Nowadays, due to its high temporal and spatial resolution, fluorescence imaging technology has become one of the most effective techniques in monitoring of the production, transport and biological functions of biomolecules in the context of life systems. Therefore, fluorescence imaging is highly important in many different research fields, such as biology, clinical diagnosis, drug discovery and development [4-6]. The development of high-sensitivity fluorescence techniques and visualizations in low-concentration fluorophore analysis has made it possible to develop a specific performance of biological interactions between different types of cells via new small molecule fluorescent compounds [7]. Up to now, a great number of small molecule fluorescent probes have been developed for sensing, imaging and biomedical applications. In particular, the fluorescent properties of small molecule fluorescent probes can be easily adjusted by synthetic chemical modification. Therefore, various types of small molecule fluorescent probes for detecting biological components (e.g., enzymes, metal ions and amino acids) have been developed [8, 9].
A fluorescent probe is composed of a fluorophore and a receptor unit [10]. A fluorescence change caused by the reaction of the receptor unit with the analyte serves as a detection effect [11], and there are several mechanisms for the fluorescence change. The most commonly applied fluorescence quenching mechanisms are photoinduced electron transfer (PET), intramolecular charge transfer (ICT) and Förster resonance energy transfer (FRET) [12]. PET refers to the electron transfer from the donor to the excited state of the fluorophore [13]. ICT is the change in fluorescence caused by electron transfer within the fluorophore molecule [14], while FRET is a quenching mechanism in which a donor fluorophore transfers the energy of its excited state to an acceptor fluorophore of a low energy excited state [15].
TP fluorescence imaging is based on the principle of simultaneously absorbing two near-infrared photons under photoexcitation [16]. Compared to traditional one-photon fluorescence imaging, TP fluorescence imaging is the use of longer excitation wavelength, resulting in higher energy to get deeper imaging depth (up to millimeter level). Therefore compared with one-photon fluorescence imaging, it has superior properties such as relatively lower cell damage and photobleaching, deeper cell penetration and weaker background fluorescence interference [17-19]. TP fluorescence imaging is widely used in in vivo imaging, and it is ideal for bioimaging and molecular probe activation. Therefore, development of TP small molecule enzyme probes is of great significance for the early diagnosis of diseases [20-22]. In order to introduce the recent advances in TP small molecule enzymatic probes, in this review, we summarize the design principles and examples of the corresponding probes that target various enzymes.
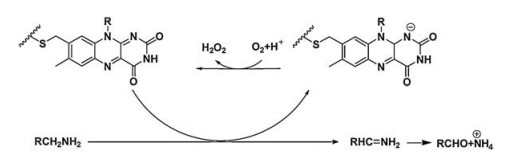
2. TP small molecule enzymatic probes for monoamine oxidasesMonoamine oxidases (MAOs) are flavin enzymes that catalyze the oxidation of monoamines, and their substrates are neurotransmitters such as dopamine, norepinephrine and serotonin. MAOs catalyze the oxidative conversion of the amine substrate to the imine under aerobic conditions, which are then hydrolyzed to the aldehyde product and releases hydrogen peroxide (Fig. 1). MAOs are expressed in the brain and most peripheral tissues; they are mainly located on the mitochondrial outer membrane of the cells. MAOs have two isoforms, MAO-A and MAO-B. Although the two flavozymes have 70% similarity in the amino acid sequence and 90% similarity in the corresponding active cavities, they have different physiological properties, substrate preferences and inhibitor sensitivities [23]. In the brain, MAO-A is mainly expressed in catecholaminergic neurons, while MAO-B is mainly expressed in serotonergic neurons, histaminergic neurons and astrocytes. MAO-B accounts for more than 80% of the total MAOs' activity in the human brain. In peripheral tissues, MAO-A predominates in fibroblasts and placental tissues, while MAO-B is more active in platelets and lymphocytes. Any excessive or deficient activity of these enzymes can lead to a variety of neurological and psychiatric disorders such as depression, PD, AD, and brain tumor [1, 24-26]. For example, AD and PD are associated with elevated levels of MAO-B in the cerebral cortex and hippocampus [27]. Therefore, the detection/imaging using TPFPs for MAOs' activity has its clinical importance in the earlydiagnosis of above diseases [1, 24-26].

|
Download:
|
| Fig. 1. Reaction mechanism of MAOs. Copied with permission [23]. Copyright 2019, Wiley Publishing Group. | |
{kind=link}
In 2012, Kim et al. [28] designed and synthesized two TPFPs (probes 1a and 1b) that were used for detecting and imaging MAOs activity in living cells (Fig. 2). Both probes 1a and 1b are derivatives of naphthalene as a fluorophore precursor, while aminopropyl and N-methylaminopropyl groups are applied as recognition groups. Upon the catalysis of MAOs, the probe in the fluorescence quenching state is converted into a product with strong fluorescence emission through three steps (i.e., oxidation reaction, elimination reaction and molecular condensation) to realize the fluorescence response to MAO. The experimental results have shown that probes 1a and 1b not only achieve rapid response to MAOs in vitro, but also have excellent cell permeability, enabling high-resolution TP imaging of highly expressed MAOs in chromaffin cells.

|
Download:
|
| Fig. 2. Structures of reactive probes 1a and 1b for MAOs and the sensing mechanism [28]. Copied with permission [28]. Copyright 2012, Royal Society of Chemistry. | |
{kind=link}
In 2016, the same group [29] reported the co-detection of MAO activity and amyloid-beta (Aβ) plaques in aging AD-bearing mice using probe 1a. Probe 1a under the catalysis of MAOs produces dipolar fluorophore that senses Aβ plaques, a general AD biomarker, enabling them to jointly study MAOs' activity and AD development. The results indicate that the progression of AD is closely related to MAOs' activity, and histological analysis revealed that MAOs' activity around Aβ plaques is elevated in aged mice. The interaction of MAOs' activity with Aβ plaques in AD mice model is demonstrated by in vivo imaging for the first time. This combination is achieved by a reaction-based TP MAOs probe of which the enzymatic reaction product can pass through the bloodbrain barrier and sense Aβ plaques to a depth of 600 μm.
Our group [30] designed and synthesized the first TP fluorescence probe 2 (Fig. 3) that could be used for the specific detection and imaging of MAO-B in the PD models. Probe 2 used 2- methylamino-6-acetylnaphthalene as the TP reporter. 3-Aminopropyl carbamate acted as a MAOs reaction unit. The carbamate with strong electron pull-down interferes the push-pull electron system of the acedan fluorophore, leaving the probe in fluorescence quenching state. Upon the recognition by MAO-B, the oxidative probe and β-elimination reactions release the acedan fluorophore under the catalysis of the enzyme therefore realizing fluorescence response to MAO-B. It enables high sensitivity, specificity and real-time imaging of endogenous MAO-B activity in biological samples. Probe 2 was used to confirm the inverse relationship between PD and MAO-B reported in the PD models. Without significant toxicity, probe 2 was applied to monitor MAO-B activity in small animals during disease progression. In clinical samples, we found that MAO-B activity did only elevate in B lymphocytes (not in fibroblast cells) suggesting that MAO-B activity in peripheral blood cells might be an available biomarker for rapid detection of PD. These results provided an important starting point for the use of small molecule imaging techniques to explore MAO-B activity at the level of organisms. However, TPFPs for targeted detection of MAO-A have not yet been developed.

|
Download:
|
| Fig. 3. (A) The mechanism by probe 2 acts on MAO-B. (B) TP fluorescence images of parkin-null drosophila. Depth (100 μm) at λex =780 nm. Copied with permission [30]. Copyright 2014, Nature Publishing Group. | |
{kind=link}
In 2015, our group [31] reported the design of the first MAO-B probe (probe 3, Fig. 4) based on bifunctional activities. The probe had high selectivity against MAO-B, and endogenous MAO-B activity can be sensitively reported by in situ proteomic analyzing/ imaging even at high MAO-A expression levels. With the built-in imaging module as part of the probe design, the probe is capable of performing live cell imaging of MAO-B activity without being affected by diffusion problems.

|
Download:
|
| Fig. 4. The design and synthesis route of probe 3. Copied with permission [31]. Copyright 2015, Wiley Publishing Group. | |
{kind=link}
3. TP small molecule enzymatic probes for nitroreductase
Nitroreductase (NTR) is a member of the enzyme containing flavin, which is found in a wide range of bacterial genera [32]. Under hypoxic conditions, overexpression of NTR has been observed and led to several malignant diseases such as rheumatoid arthritis [33], ischemic stroke [34] and solid tumor [35]. In solid tumors, the activity of NTR is directly related to the degree of hypoxia [36]. In hypoxic cells, in the case of reduction of nicotinamide adenine dinucleotide (NADH), NTR can reduce the nitroaromatic compound to the corresponding amine [37]. Therefore, NTR is used as an important indicator of the degree of hypoxia in living cells [36]. Nitroaromatic compounds have been proven to be excellent substrates for NTR, which are particularly pronounced in tumor cells [38]. Therefore, selective detection of NTR is considered to be an effective tool for tumor diagnosis [39]. On this basis, the development of a simple and effective method for detecting NTR activity is of great significance for improving the anticancer therapeutic effect. In recent years, many small molecule fluorescent probes specific to NTR have been reported. More importantly, these probes also suggest several important mechanisms and principles related to the NTR-catalyzed reduction reaction.
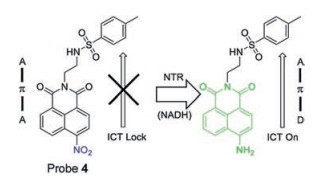
Lin's group [40] reported the first TPFP probe 4 (Fig. 5) for the detection of NTR in the endoplasmic reticulum of cells. This probe was constructed using naphthalimide as a fluorescent reporter and a nitro group as a reactive group of NTR, and a methylsulfonamide group as an endoplasmic reticulum (ER) targeting group. The nitro group quenches the fluorescence of 1, 8-naphthalimide, while the fluorescence restores at 543 nm when the nitro group is reduced by NTR, as ICT effect of the molecule results in strong fluorescence.

|
Download:
|
| Fig. 5. Structure of fluorescent NTR Probe 4. Copied with permission [40]. Copyright 2018, Elsevier Ltd. | |
{kind=link}
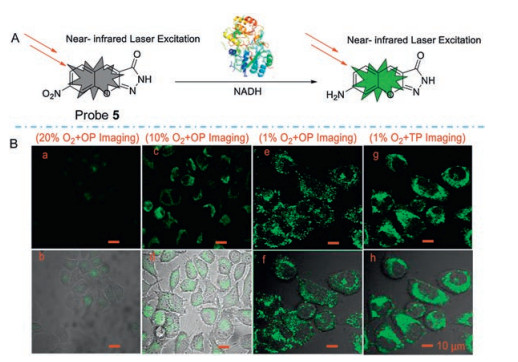
Hu and his colleagues [41] developed a novel TP fluorophore Probe 5 (Fig. 6) for detecting NTR in living tissue. After incubating with NTR for 15 min, probe 5 showed approximately 90-fold fluorescence enhancement at 505 nm and a maximum TP action cross-section at 760 nm of 200 GM. They also demonstrated that the probe remains its high fluorescence enhancement at the tissue level (50–165 μm). Zhang's group [42] developed a TPFP (probe 6, Fig. 7) for detecting NTR in cancer stem cells. The TP fluorescence excitation spectrum of probe 6 extends to the near-infrared (NIR) region, therefore significantly improve the tissue penetration ability. This probe has the potential to be used in early diagnose of cancers.

|
Download:
|
| Fig. 6. (A) The mechanism of fluorescent NTR Probe 5. (B) OP and TP images of Hela cell under normoxic and different hypoxia conditions. OP: λex =405 nm; TP: λex =760 nm. λem: 490–530 nm. Copied with permission [41]. Copyright 2018, Elsevier Ltd. | |
{kind=link}

|
Download:
|
| Fig. 7. The mechanism of fluorescent NTR probe 6. Copied with permission [42]. Copyright 2018, Elsevier Ltd. | |
{kind=link}
4. TP small molecule enzymatic probes for γ-glutamyltranspeptidase
γ-Glutamyltranspeptidase (GGT) is a cell membrane-bound protease involved in the homeostasis of glutathione (Glu) and cysteine (Cys). It is ubiquitous in mammalian cells/bacteria and is closely related to many important physiological processes. Abnormal levels of GGT can be considered useful biomarkers for various diseases such as hepatotoxicity, asthma, diabetes and bacterial infections. In addition, recent evidence suggests that GGT is overexpressed in many malignancies, such as cervical and breast cancer [32, 43-46].
In 2016 [47], Wu and his group, for the first time, have reported a TPFP for GGT (probe 7 in Fig. 8), that is able to detect GTT in zebrafishes. This probe is consisted of a dicyanomethylene-4H-pyranyl group (DCM) and a γ-glutamine. In the presence of GTT, the γ-glutamine moiety of the probe is cleaved to restore fluorescence emission at 635 nm under a femtosecond pulse of 800 nm.

|
Download:
|
| Fig. 8. The structures of probes of GGT and the typical mechanism (below). Reproduced with permission [47-49]. Copyright 2015, Elsevier Ltd. [47]; Copyright 2015, Royal Society of Chemistry [48]. Copyright 2018, Elsevier Ltd. [49]. | |
{kind=link}
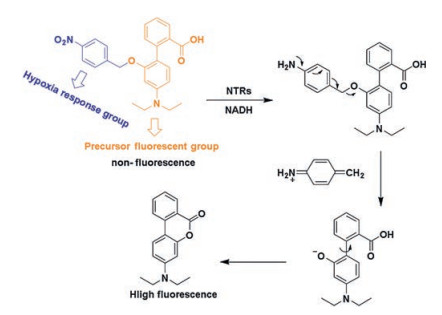
Next, Zhang's group [48] reported a novel TP fluorescent conduction probe (probe 8 in Fig. 8), which is designed based on naphthalene derivatives with gamma-glutamyl as a recognition group for GGT detection and bioimaging. After excitation at 780 nm, images were collected at 450–550 nm with femtosecond pulses. This probe restored strong fluorescence emission with the catalysis of GGT. Moreover, the depth of tissue imaging of rat liver cancer is 70–160 μm.
Peng et al. [49] developed a 2-dicyanomethylidene-3-cyano- 4, 5, 5-trimethyl-2, 5-ihydrofuran (TCF) derivative for GGT detection in tumor cells (probe 9 in Fig. 8). The probe is the first "double" labeled 2D and 3D fluorescent imaging probe that is able to identify A2780 cells in a mixed culture dish. Moreover, probe 9 can detect endogenous GGT activity in zebrafishes, which has great potential in GTT inhibitors screening.
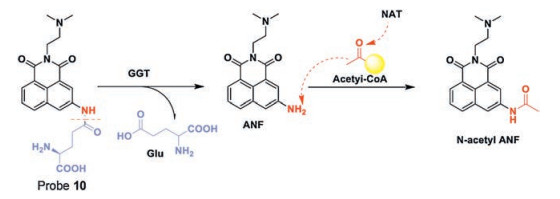
Guo's group [50] developed a TPFP (probe 10, Fig. 9) derived from naphthalimide based on a sequential ICT process for the detection of GGT and N-acetyltransferase (NAT). First, at the cell surface, GGT cleaves the Glu group from the probe, therefore the first ICT process occurs, and the blue fluorescence turns green. Then, when the probe is in the DNA damage region, NAT transfers an acetyl group to the naphthalimide with the existence of acetylcoA, therefore the second ICT process occurs, and restores the blue fluorescence of probe 10. Hence, imaging DNA damage in cancer cells is performed based on the fluorescence emission of the probe that changes from blue to green, then back to blue fluorescence again. More importantly, by observing the apparent and high signal-to-background fluorescence from blue to green and returning to blue under TP excitation, the real-time DNA damage in cancer cells and intact animals can be observed by naked-eye and handheld fluorescent light emitted at 365 nm. Dynamic imaging can also be observed using a TP fluorescence microscope. In summary, this sequential ICT-reactive fluorescent probe is promising as a visualization tool for real-time dynamic imaging of DNA damage.

|
Download:
|
| Fig. 9. Structure of fluorescent GGT probe 10. Reproduced with permission [50]. Copyright 2016, Royal Society of Chemistry. | |
{kind=link}
5. TP small molecule enzymatic probes for phosphatase
Phosphatases are a large family of enzymes [1], including protein tyrosine phosphatases (PTPs) [51], alkaline phosphatases (ALPs) [52] and acid phosphatases [53]. The biological functions of these enzymes are distinct. PTPs play an important role in the tyrosine phosphorylation of substrates and participate in the process of microtubule nucleation [54]. ALPs play crucial part in adjusting biological processes involved in signal transduction pathways, growth, apoptosis, and cell cycle in living organisms [52]. The levels of acid phosphatases are associated with Gaucher disease, and diseases associated with kidneys, veins and bones [55].
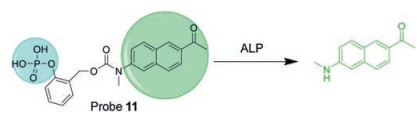
Sun and his team reported a probe that is consisted of a TP fluorophore [56], a phosphate recognition moiety and a selfcleaving adaptor (probe 11, Fig. 10). It has fast reaction kinetics, high sensitivity and low cytotoxicity, and it has higher selectivity against ALP. Probe 11 has been shown to be suitable for the detection of endogenous ALP activity in living cells and fresh rat tissues using confocal microscopy, with imaging depths up to 140 μm. However, this probe is not suitable for in vivo imaging due to its green fluorescence emission.

|
Download:
|
| Fig. 10. Chemical structure of TP-Phos probe 11 and its proposed "turn-on" mechanism by ALP. Copied with permission [56]. Copyright 2017, Elsevier Ltd. | |
{kind=link}
Our group reported a TP probe 12 (Fig. 11) for phosphatase detection [57-59], which can "fix" the target phosphatase with the fluorescent probe via covalently binding to the enzyme. By using an appropriate linker and mandelic acid core, three modular components (including a photolabile-group-caged enzyme recognition group, a fluorescent reporter, and a quencher) were strategically built around it. Upon the UV irradiation, caging group is removed and presents the naked phosphate group to the PTPs. Then the quencher is removed by 1, 6-elimination to restore a distinct fluorescence, as well as to covalently label the neighboring nucleophiles of the enzyme active site thus indicating the localization of enzymes. However, the biggest drawback of the probe is the poor cell permeability.

|
Download:
|
| Fig. 11. Probe for phosphatase. Copied with permission [57-59]. Copyright 2012, American Chemical Society [57]. Copyright 2013, Wiley Publishing Group [58]. Copyright 2011, American Chemical Society [59]. | |
{kind=link}
We further constructed phosphatase probe 13 (Fig. 11) [58], which were optimized with subcellular targeting peptides for specific delivery and enhanced subcellular retention with less interference compared to protein activities than probe 12. By the conjugation of phosphatase probes to different cell-penetrating peptides, the probes were able to achieve organelle- or tumor-cell-specific TP imaging of phosphatase activity with decent spatial and temporal resolutions. These probes were successfully applied for TP imaging of endogenous phosphatase activity in drosophila brains with an imaging depth of >100 μm. However, this design also suffers from a drawback in which probe 13 cannot distinguish between different classes of phosphatases, and the imaging results may be due to their different endogenous phosphatases. Thus, the probe can be activated elsewhere before it is driven to the targeted organelle by the cell-penetrating peptides (CPPs).
In order to address the above issue, we developed a membranetargeted fluorescent probe 14 (Fig. 11) for TP imaging of membrane associated phosphatase activity [59]. The probe contains a cell membrane anchored on a TP fluorophore and quencher linked 'photocaged' phosphate moiety. These two parts are combined together through electrostatic interactions; thereby effectively quench the fluorescence through intermolecular FRET. Subsequently, upon UV irradiation, the deprotected phosphate can undergo enzymatic dephosphorylation by membrane associated phosphatase, thus leads to the dissociation of the quencher from the fluorophore and restores its fluorescence. Importantly, these probe-designing strategies resolve the diffusion problem, which is typically associated with most small-molecule fluorescent probes, affording the capability of in situ imaging. However, the covalentbinding probe may interfere with the enzymatic activity.
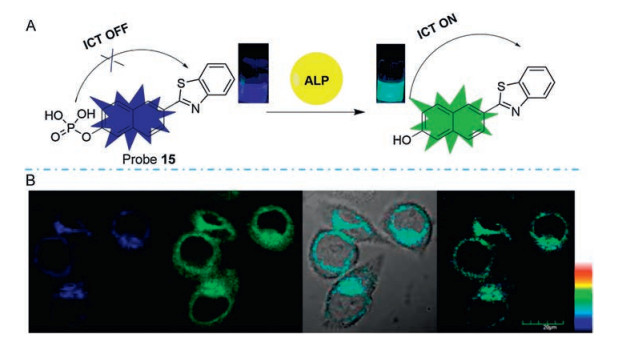
Zhu [60] and his group reported an efficient TP ratiometric fluorescent probe (probe 15) that is designed based on classic naphthalene derivatives with a donor-π-acceptor (D-π-A) structure (Fig. 12). The presence of ALP causes the cleavage of the phosphate group from naphthalene derivatives and the phosphate group changes the ability of the ICT and remarkably alters the probe's photophysical properties, thus an obvious ratiometric signal with an isoemissive point is observed. The fluorescence intensity ratio displayed a linear relationship against the concentration of ALP in the concentration range from 20 U/L to 180 U/L with the limit of detection of 2.3 U/L. Additionally, the probe 15 is further used for fluorescence imaging of ALP in living cells under one-photon (405 nm) or TP excitation (720 nm) with high resolution images that demonstrates the potential of probe 15 in various biological applications.

|
Download:
|
| Fig. 12. (A) Structure of TP ratiometric probe 15 and its detection mechanism toward ALP. (B) TP fluorescent imaging of HeLa cells treated with probe 15, λex =720 nm. Copied with permission [60]. Copyright 2014, Multidisciplinary Digital Publishing Institute. | |
{kind=link}
6. TP small molecule enzymatic probes for protease
Proteases are enzymes that hydrolyze proteins. They can be divided into serine proteases, thiol proteases, aspartic proteases and metalloproteinases depending on their active centers [61]. It has been found that protease activity and abnormal expression are associated with diseases such as placental dysplasia, pre-eclampsia [62], cardiovascular disease, inflammation, and cancer [63]. Notably, among different proteases, cell-surface proteases particularly play important role in physiological and pathological functions [64], therefore real-time imaging of cell-surfaceassociated proteolytic enzymes is critical to the better understanding of their activities in both physiological and pathological processes [1].
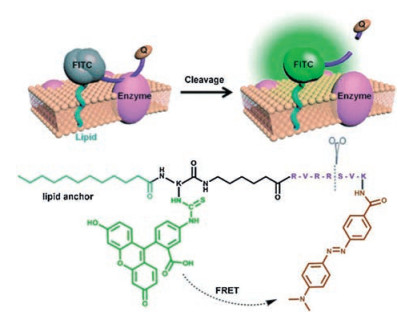
Xing and co-workers have designed and synthesized a unique small-molecule fluorescent probe 16 [65], which combines the principles of passive exogenous membrane insertion and FRET to image cell-surface-localized furin-like convertase activities. As shown in the Fig. 13, the membrane-associated furin-like enzymatic cleavage of the peptide probe leads to an increased fluorescence intensity which was mainly localized on the plasma membrane of the furin-expressed cells. This small-molecule fluorescent probe could serve as a unique and reliable reporter for real-time visualization of endogenous cell-surface associated proteolytic furin-like enzyme functions in live cells and tissues using one-photon and TP fluorescence microscopy.

|
Download:
|
| Fig. 13. The structure and detection mechanism of probe 16. Copied with permission [65]. Copyright 2014, Wiley Publishing Group. | |
{kind=link}
7. Conclusion and outlook
Nowadays, a series of TP small molecule fluorescent probes for enzymes have been developed which have good prospects in the clinic. While making these advances, to the best of our knowledge, there are still several physiologically and pathologically important enzymes that do not have corresponding TPFPs, such as acylprotein thioesterases (APTs) that are important in lipid metabolism [66, 67], glycosidases which is closely associated with diabetes [68-70], thioredoxin reductase (TrxR) and thioredoxin (Trx) that are linked with cardiovascular disease [71, 72] and cellular antioxidants [73] respectively. Therefore, we look forward to the development specific small-molecule fluorescent probes in the detection of other enzymes. In addition, higher selectivity to detect a particular subtype, lower biotoxicity to improve clinical possibilities, deeper tissue penetration depth, and faster response time will also be the main direction of future optimization. At the same time, we also expect that the probe has decent water solubility and biocompatibility. Lastly, on the basis of the existing probes, higher fluorescence quantum yield, better luminescence performance and lower autofluorescence in TPFPs are also desired. In conclusion, TPFPs holds great potentials in the rapid detection of enzyme activity and early diagnosis of disease.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 81672508), Jiangsu Provincial Foundation for Distinguished Young Scholars (No. BK20170041), Natural Science Foundation of Shaanxi Province (No. 2019JM-016), China-Sweden Joint Mobility Project (No. 51811530018) and Fundamental Research Funds for the Central Universities.
| [1] |
L. Qian, L. Li, S.Q. Yao, Acc. Chem. Res. 49 (2016) 626-634. DOI:10.1021/acs.accounts.5b00512 |
| [2] |
H.M. Kim, B.R. Cho, Chem. Rev. 115 (2015) 5014-5055. DOI:10.1021/cr5004425 |
| [3] |
H. Wei, G. Wu, X. Tian, et al., Future Med. Chem. 10 (2018) 2729-2744. DOI:10.4155/fmc-2018-0193 |
| [4] |
A.B. Chinen, C.M. Guan, J.R. Ferrer, et al., Chem. Rev. 115 (2015) 10530-10574. DOI:10.1021/acs.chemrev.5b00321 |
| [5] |
L. Yuan, W. Lin, K. Zheng, et al., Chem. Soc. Rev. 42 (2013) 622-661. DOI:10.1039/C2CS35313J |
| [6] |
L. Wang, W. Du, Z. Hu, et al., Angew. Chem. Int. Ed. 58 (2019) 2-20. DOI:10.1002/anie.201813331 |
| [7] |
B.A. Neto, P.H. Carvalho, J.R. Correa, Acc. Chem. Res. 48 (2015) 1560-1569. DOI:10.1021/ar500468p |
| [8] |
H. Kobayashi, P.L. Choyke, Acc. Chem. Res. 44 (2011) 83-90. DOI:10.1021/ar1000633 |
| [9] |
T. Kowada, H. Maeda, K. Kikuchi, Chem. Soc. Rev. 44 (2015) 4953-4972. DOI:10.1039/C5CS00030K |
| [10] |
X.H. Li, X.H. Gao, W. Shi, et al., Chem. Rev. 114 (2014) 590-659. DOI:10.1021/cr300508p |
| [11] |
M. Kaur, D.H. Choi, Chem. Soc. Rev. 44 (2015) 58-77. DOI:10.1039/C4CS00248B |
| [12] |
Y. Fu, N.S. Finney, RSC Adv. 8 (2018) 29051-29061. DOI:10.1039/C8RA02297F |
| [13] |
T.I. Kim, D. Kim, J. Bouffard, et al., Sens. Actuators B Chem. 283 (2019) 458-462. DOI:10.1016/j.snb.2018.12.017 |
| [14] |
C. Zhang, Z. Han, M. Wang, et al., Dalton Trans. 47 (2018) 2285-2291. DOI:10.1039/C7DT04345G |
| [15] |
Y.L. Wu, S.L. Huang, F. Zeng, et al., Chem. Commun. 51 (2015) 12791-12794. DOI:10.1039/C5CC04771D |
| [16] |
J. Morales-Dalmau, C. Vilches, V. Sanz, et al., Nanoscale 11 (2019) 11331-11339. DOI:10.1039/C9NR01198F |
| [17] |
X. Lou, Z. Zhao, B.Z. Tang, Small 12 (2016) 6430-6450. DOI:10.1002/smll.201600872 |
| [18] |
M.K. Kim, C.S. Lim, J.T. Hong, et al., Angew. Chem. Int. Ed. 49 (2010) 364-367. DOI:10.1002/anie.200904835 |
| [19] |
B.K. Agrawalla, Y. Chandran, W.H. Phue, et al., J. Am. Chem. Soc. 137 (2015) 5355-5362. DOI:10.1021/ja5115776 |
| [20] |
H.M. Kim, B.R. Cho, Acc. Chem. Res. 42 (2009) 863-872. DOI:10.1021/ar800185u |
| [21] |
G. Bort, T. Gallavardin, D. Ogden, et al., Angew. Chem. Int. Ed. 52 (2013) 4526-4537. DOI:10.1002/anie.201204203 |
| [22] |
M.D. Cahalan, I. Parker, S.H. Wei, et al., Nat. Rev. Immunol. 2 (2002) 872-880. DOI:10.1038/nri935 |
| [23] |
R. Shi, Q. Wu, C. Xin, et al., ChemBioChem 20 (2019) 1487-1497. |
| [24] |
L.G. Iacovino, F. Magnani, C. Binda, J. Neural Transm. 125 (2018) 1567-1579. DOI:10.1007/s00702-018-1915-z |
| [25] |
M. Naoi, P. Riederer, W. Maruyama, J. Neural Transm. 123 (2016) 91-106. DOI:10.1007/s00702-014-1362-4 |
| [26] |
K.F. Tipton, J. Neural Transm. 125 (2018) 1519-1551. DOI:10.1007/s00702-018-1881-5 |
| [27] |
J.C. Wang, C.L. Cheng, C. Xin, et al., Molecules 24 (2019) 16. |
| [28] |
D. Kim, S. Sambasivan, H. Nam, et al., Chem. Commun. 48 (2012) 6833-6835. DOI:10.1039/c2cc32424e |
| [29] |
D. Kim, S.H. Baik, S. Kang, et al., ACS Cent. Sci. 2 (2016) 967-975. DOI:10.1021/acscentsci.6b00309 |
| [30] |
L. Li, C.W. Zhang, G.Y. Chen, et al., Nat. Commun. 5 (2014) 3276. DOI:10.1038/ncomms4276 |
| [31] |
L. Li, C.W. Zhang, J. Ge, et al., Angew. Chem. Int. Ed. 54 (2015) 10821-10825. DOI:10.1002/anie.201504441 |
| [32] |
W. Qin, C. Xu, Y. Zhao, et al., Chin. Chem. Lett. 29 (2018) 1451-1455. DOI:10.1016/j.cclet.2018.04.007 |
| [33] |
L. Lelieveldt, H. Kristyanto, G.J.M. Pruijn, et al., Mol. Pharm. 15 (2018) 5565-5573. DOI:10.1021/acs.molpharmaceut.8b00741 |
| [34] |
J.R. Zheng, Y.Z. Shen, Z.Q. Xu, et al., Biosens. Bioelectron. 119 (2018) 141-148. DOI:10.1016/j.bios.2018.08.014 |
| [35] |
Z.W. Liu, F.L. Song, W.B. Shi, et al., ACS Appl. Mater. Interfaces 11 (2019) 15426-15435. DOI:10.1021/acsami.9b04488 |
| [36] |
H.W. Liu, L. Chen, C. Xu, et al., Chem. Soc. Rev. 47 (2018) 7140-7180. DOI:10.1039/C7CS00862G |
| [37] |
Y. Fang, W. Shi, Y. Hu, et al., Chem. Commun. 54 (2018) 5454-5457. DOI:10.1039/C8CC02209G |
| [38] |
F. Song, R. Liang, J. Deng, et al., Chin. Chem. Lett. 28 (2017) 1997-2000. DOI:10.1016/j.cclet.2017.08.023 |
| [39] |
S.Y. Na, S. Park, S.Y. Kim, et al., Dyes Pigm. 161 (2019) 247-251. DOI:10.1016/j.dyepig.2018.09.053 |
| [40] |
A. Xu, Y. Tang, W. Lin, Spectrochim. Acta Part A 204 (2018) 770-776. DOI:10.1016/j.saa.2018.05.092 |
| [41] |
L. Zhou, L. Gong, S. Hu, Spectrochim. Acta Part A 199 (2018) 254-259. DOI:10.1016/j.saa.2018.03.073 |
| [42] |
Y. Liu, W. Liu, H. Li, et al., Anal. Chim. Acta 1024 (2018) 177-186. DOI:10.1016/j.aca.2018.03.030 |
| [43] |
C. Wang, X. Song, L. Chen, et al., Biosens. Bioelectron. 91 (2017) 313-320. DOI:10.1016/j.bios.2016.11.018 |
| [44] |
W. Liu, B. Huang, Z.X. Tong, et al., New J. Chem. 42 (2018) 5403-5407. DOI:10.1039/C8NJ00520F |
| [45] |
L. Feng, P. Li, J. Hou, et al., Anal. Chem. 90 (2018) 13341-13347. DOI:10.1021/acs.analchem.8b02857 |
| [46] |
J. Zhang, Z. Jin, X.X. Hu, et al., Anal. Chem. 89 (2017) 8097-8103. DOI:10.1021/acs.analchem.7b01659 |
| [47] |
P. Zhang, X.F. Jiang, X. Nie, et al., Biomaterials 80 (2016) 46-56. DOI:10.1016/j.biomaterials.2015.11.047 |
| [48] |
P. Wang, J. Zhang, H.W. Liu, et al., Analyst (Cambridge.. U. K.) 142 (2017) 1813-1820. DOI:10.1039/C7AN00229G |
| [49] |
H. Li, Q. Yao, F. Xu, et al., Biomaterials 179 (2018) 1-14. DOI:10.1016/j.biomaterials.2018.06.028 |
| [50] |
H. Zhang, K. Wang, X. Xuan, et al., Chem. Commun. 52 (2016) 6308-6311. DOI:10.1039/C6CC02290A |
| [51] |
T. Durgannavar, S.J. Kwon, A.B.T. Ghisaidoobe, et al., ChemBioChem 19 (2018) 2495-2501. DOI:10.1002/cbic.201800529 |
| [52] |
Q. Huang, C. He, J. Zhang, et al., Anal. Chim. Acta 1055 (2019) 98-105. DOI:10.1016/j.aca.2018.12.035 |
| [53] |
X.X. Li, B.Z. Li, J.L. Hong, et al., Sens. Actuators B:Chem. 276 (2018) 421-428. DOI:10.1016/j.snb.2018.08.076 |
| [54] |
A. Klebanovych, V. Sladkova, T. Sulimenko, et al., Cells 8 (2019) 345. DOI:10.3390/cells8040345 |
| [55] |
Z. Fredj, M.B. Ali, M.N. Abbas, et al., Anal. Chim. Acta 1057 (2019) 98-105. |
| [56] |
H. Zhang, P. Xiao, Y.T. Wong, et al., Biomaterials 140 (2017) 220-229. DOI:10.1016/j.biomaterials.2017.06.032 |
| [57] |
M.Y. Hu, L. Li, H. Wu, et al., J. Am. Chem. Soc. 133 (2011) 12009-12020. DOI:10.1021/ja200808y |
| [58] |
L. Li, J. Ge, H. Wu, et al., J. Am. Chem. Soc. 134 (2012) 12157-12167. DOI:10.1021/ja3036256 |
| [59] |
L. Li, X. Shen, Q.H. Xu, et al., Angew. Chem. Int. Ed. 52 (2013) 424-428. DOI:10.1002/anie.201205940 |
| [60] |
X. Zhou, Y. Jiang, X. Zhao, et al., Molecules 21 (2016) 1619. DOI:10.3390/molecules21121619 |
| [61] |
S. Paschkowsky, J.M. Hsiao, J.C. Young, et al., Biochem. Cell Biol. 97 (2019) 265-269. DOI:10.1139/bcb-2018-0186 |
| [62] |
A. Inagaki, H. Nishizawa, S. Ota, et al., Placenta 33 (2012) 919-926. DOI:10.1016/j.placenta.2012.08.003 |
| [63] |
B. Turk, Nat. Rev. Drug Discov. 5 (2006) 785-799. DOI:10.1038/nrd2092 |
| [64] |
M.S. Brown, J. Ye, R.B. Rawson, et al., Cell 100 (2000) 391-398. DOI:10.1016/S0092-8674(00)80675-3 |
| [65] |
J. Mu, F. Liu, M.S. Rajab, et al., Angew. Chem. Int. Ed. 53 (2014) 14357-14362. DOI:10.1002/anie.201407182 |
| [66] |
M.W. Beck, R.S. Kathayat, C.M. Cham, et al., Chem. Sci. 8 (2017) 7588-7592. DOI:10.1039/C7SC02805A |
| [67] |
R.S. Kathayat, Y. Cao, P.D. Elvira, et al., Nat. Commun. 9 (2018) 334. DOI:10.1038/s41467-017-02655-1 |
| [68] |
L. Chao, S.A.K. Jongkees, Angew. Chem. Int. Ed. 58 (2019) 2-13. DOI:10.1002/anie.201813331 |
| [69] |
D. Li, L. Liang, Y. Tang, et al., Chin. Chem. Lett. 30 (2019) 1013-1016. DOI:10.1016/j.cclet.2018.12.022 |
| [70] |
J.J. Li, K.R. Wang, R.F. Li, et al., J. Mater. Chem. B 7 (2019) 1270-1275. DOI:10.1039/C8TB03122C |
| [71] |
Y. Liu, H. Ma, L. Zhang, et al., Chem. Commun. 52 (2016) 2296-2299. DOI:10.1039/C5CC09998F |
| [72] |
X. Li, B. Zhang, C. Yan, et al., Nat. Commun. 10 (2019) 2745. DOI:10.1038/s41467-019-10807-8 |
| [73] |
M.H. Lee, H.M. Jeon, J.H. Han, et al., J. Am. Chem. Soc. 136 (2014) 8430-8437. DOI:10.1021/ja503356q |