 2019, Vol. 30
2019, Vol. 30 


b Department of Organic Chemistry, College of Pharmacy, Second Military Medical University, Shanghai 200433, China
Fluorescent imaging is one of the most crucial techniques in revealing complicated biological structures and processes in living cells. With the rapid innovation in microscopy techniques recently, super-resolution fluorescence microscopy such as stimulated emission depletion microscopy (STED), stochastical optical resolution microscopy (STORM), structure illumination microscopy (SIM) et al., have allowed us to unveil fine biological structures and bioactivities beyond the diffraction limit in vivo [1-5]. To achieve desired temporal and spatial resolution with such sophisticated microscopy, the design and construction of fluorophores for specific systems become indispensable. Fluorescent probes with distinct photophysical and photochemical properties, including absorption and emission wavelength, fluorescent quantum yield, fluorescent intensity, stability and reactivity with specific targets, are urgently demanded.
Organic fluorophores, fluorescent proteins, quantum dots and lanthanide chelates have been applied as the fluorescent probes in fluorescent imaging. Organic fluorophore, which belongs to an ancient area that dates back to the beginning of fluorescent imaging, has attracted growing attention recently. Fluorescent probes based on xanthene ring structure are particularly attractive with excellent photochemical properties, including high molar absorption coefficient, high fluorescence quantum yield, outstanding water solubility and photostability. Rhodamine, an exemplary case, has been widely used as a platform for the develop of novel fluorophores [6, 7]. However, the emission of rhodamines mostly falls into ultraviolet or visible region, which is susceptible to the background fluorescence in the biological samples. Moreover, fluorescent at visible range usually got attenuated in biological sample due to scattering from tissues. Fluorophores in nearinfrared (NIR, 650 - 900 nm) range that offer strong penetration and low phototoxicity become favored for fluorescent imaging with high signal to noise ratio in complicated biological samples. Recently, NIR fluorophores that inherit the merit of traditional rhodamine frameworks have been extensively explored for the construction of fluorescent probes.
In 2008, Xiao and Qian et al. [8-10] have pioneeringly reported that the substitution of bridging oxygen atom by silicon atoms in rhodamine framework yields strong fluorophore with bathochromic shifted spectra (90 nm) and high molar extinction coefficient [11]. This is the first attempt of introducing heteroatom silicon into the skeleton of rhodamines for tuning their optical properties. Inspired by this design concept, Nagano's group [12-14] and other pioneer groups in the area [15-18] have proved that Si-substituted rhodamine with outstanding photophysical properties is versatile for the design of exceptional NIR fluorophores in the investigation of biological systems. Except for silicon, a number of other main group elements, including P [19-22], B [23], S [24], C [25-28], Ge [12], Se [29], Sn [20] and Te [30, 31] have also provided fluorophores with tuned photophysical properties through substitution of bridging oxygen in rhodamine. Substitution of bridging oxygen in xanthene structure has been recognized as a distinctive route for the construction of novel fluorophores. However, in consideration of their excellent properties, these fluorophores are still extraordinarily under exploration, one of the most crucial obstacles is their laborious and expensive synthetic processes [32, 33]. Thus, thoughtful understanding in the synthesis and distinctive properties of substituted rhodamines might bring some new ideas into this area, which will surely sparkle the application of these fluorophores in fluorescent imaging. Some excellent review papers have extensively discussed the synthesis of Sisubstituted xanthene fluorophores, especially [34-36]. Herein, we are going to review some recent progress in the synthesis and unique properties of several representative fluorphores: silicon, phosphorus and other elements substituted rhodamines.
2. Synthesis of substituted rhodamineTetramethylrhodamine, the first rhodamine, was successfully synthesized in 1887 by Swiss chemist Maurice Ceresole through heating the mixture of 3-aminophenol and isobenzofuran-1, 3- dione in the existence of Brønsted or Lewis acid [37, 38]. Recently, some groups have also contributed to the efficient synthesis of rhodamines with innovated procedures [39, 40]. However, the synthetic routes are not applicable in substituted rhodamines. Several approaches are now available for the synthesis of substituted rhodamines. Herein, we are going to discuss three reported synthetic routes for substituted rhodamine with the most extensively studied Si-substituted rhodamine as the example.
One of the most widely employed synthetic approach for Sirhodamine based on the nucleophilic addition: the dibromoides give an important intermediate, Si-xanthone, which was added metalated aryl species for the Si-rhodamine scaffold (Fig. 1, Route A) [41]. Other than Si-rhodamines, carbon, phosphorus [19], selenium [12], germanium [12], sulfur [24] and tellurium [30, 31] substituted rhodamines are also achievable through this route. In this process, Si-xanthone is the key intermediate, which is also one of the most challenging part in the synthesis of rhodamines. Generally, KMnO4 or quinones are used for the oxidation of rhodaminepyronines into xanthene, however, the yield is usually low. Anslyn and coworkers have recently reported the improvement in the synthesis of O-, Si- and C-xanthone without the application of harsh oxidants [42]. They succeeded in direct oxidation of the corresponding rhodaminepyronines by I2.

|
Download:
|
| Fig. 1. Three synthetic approaches for Si-rhodamines. | |
This nucleophilic addition approach is one of the most extensively used and classical way for the synthesis of substituted rhodamines. Although this procedure is versatile, it is still suffered from relative low yield and multi-step synthesis. Especially for the synthesis of some derivatives, complicated protection and deprotection are required due to the usage of active lithium agents.
To overcome the synthetic difficulties in substituted rhodamines, we have reported the synthesis of rhodamine through a condensation process with relative high yield: the dibromoides gave diaryl substituted ether intermediates, which further reacted with benzaldehydes by condensation (Fig. 1, Route B) [43]. This reaction is feasible for the synthesis of Si-rhodamines with various substituents in the absence of protection, including —Br, —COOH, —NH2, —CN and —C≡CH. However, the condensation process is still suffered from the relative tough reaction condition.
Researchers have recently reported the synthesis of Si-rhodamines by Route C (Fig. 1, Route C) [44, 45]. Rather than the nucleophile addition of metalated aryl species to Si-anthrone as Route A, they have succeeded in electrophilic reaction of bis(2- bromophenyl)silane and anhydride (or ester) for Si-rhodamines. Although protection is still required due to the usage of reactive lithium agents, many Si-rhodamines with various substitutions can be synthesized with relative high yield within 3–5 steps. One of the most exciting examples, the previous inaccessible rhodamines with tetrafluorination of the bottom aryl rings and replacement of standard N, N-dialkylamino moieties with four-membered azetidine rings can be readily synthesized. Innovation in the efficient and feasible synthesis of synthetic approach for substituted rhodamine will surely further boom their applications in many aspects.
3. Unique properties of substituted rhodaminesAfter substitution of the endocyclic oxygen atom with other elements, the π-conjugated skeletons of xanthene structures got significantly perturbed. The affected electronic structures of substituted rhodamines resulted in distinctive physicochemical characteristics, including shift in fluorescent spectra, change of chemical stability, variation in solubility, molar absorption, fluorescent quantum yield, etc. These unique properties of substituted rhodamines have flourished various fluorophores and enriched the design strategy of fluorescent probes. In this brief review, the shift in optical properties and the effects on two important characteristics that related to fluorescence controlling mechanisms: equilibrium of the spirolactone and zwitterion, multiphoton absorption and photoinduced electron transfer (PeT) efficiency of the fluorophores are discussed in detail for the substituted rhodamines.
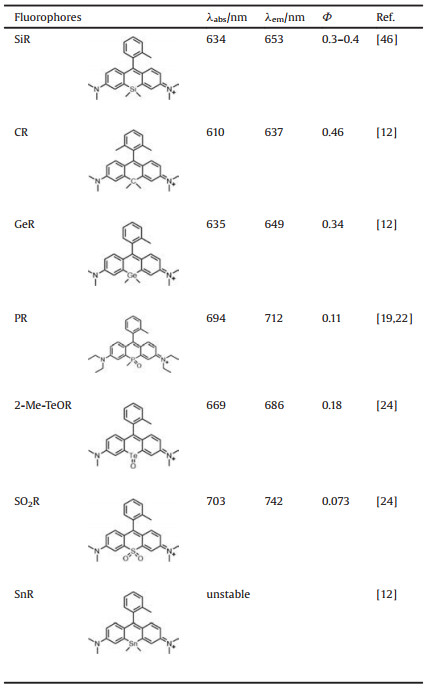
3.1. Optical propertiesIn Si substituted rhodamines (SiR for short), the significantly red-shift in fluorescence (about 90 nm compared with their mother structures) into NIR region have initiated a burst of enthusiasm in SiR. As a qualified fluorescent skeleton, SiR have offered bright fluorescence in NIR region with inherited merit of rhodamine structures, facilitating the fluorescent imaging of biological structures and bioactivities. The bathochromic shift of spectra for the fluorophores have been well explained by the alternation of their electronic structures after different substitution. The electronic properties are usually evaluated by density function theory (DFT) and time dependent (TD)-DFT calculation in theoretical calculation. In SiR, the shift of spectra agreed with the shrinking of the gap between highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). Due to the σ*-π* conjugation resulted from interaction of the σ* orbital of the two exocyclic bonds on the substituted Si atom with the π* system of the butadiene part of the fluorophore, the LUMO level got significantly decreased, resulting in a bathochromic shift of the excitation and emission spectra. The fluorescence quantum yield of SiR is about 0.3-0.4, which can be increased into higher than 0.5 with the replacement of the N, N-dialkylamino group with fourmembered azetidine rings [46].
Before SiR, carbon atom was introduced into rhodamine system to substitute the bridging oxygen, forming carbon rhodamine (CR), whose fluorescence emission wavelength was significantly redshifted compared with traditional rhodamine, reaching about 640 nm with relative high fluorescence quantum yield (about 0.5). Rhodamine substituted with germanium and tin were further designed and synthesized [12]. It is likely that due to the similar σ*-π* conjugation in SiR as we discussed before, the LUMO energy levels got decreased in germanium-substituted rhodamine (GeR). GeR shows superior NIR spectral characteristics (λabs = 635 nm, λem = 649 nm) with high fluorescence quantum yield (ΦF = 0.34). The spectra of Sn-rhodamine (SnR) were not available because it was prone to decompose. However, both Geand Sn-substituted pyronine has red-shifted the absorption and emission compared with original pyronine [12], thus it is likely that the spectra of SnR might also be bathochromic shifted compared with ordinary rhodamine.
In P-substituted rhodamine (PR), the incorporation of a P=O group has further bathochromic shifted the fluorescence (Fig. 2) [19, 22]. The intramolecular charge transfer between the conjugated p system and the P-oxide moiety has stabilized the excited state of PR and lowered its LUMO level. Moreover, the σ*-π* conjugation is likely further lowered the LUMO level and resulted further bathochromic shifted spectra compared with rhodamine and SiR. The fluorescence emission wavelength of PR can reach about 710 nm with relative high fluorescence quantum yield (about 0.4) and molar absorption coefficient (ε > 50, 000 L mol-1 cm-1). These characteristics ensure PR a very suitable NIR fluorophore for bioimaging in vivo.

|
Download:
|
| Fig. 2. Computed frontier orbitals and energy levels. Copied with permission [19].Copyright 2015, Wiley. | |
Group 16 elements, including sulfur, selenium and antimony were also introduced into rhodamine. As we expected, with the absence of σ*-π* conjugation in substituted rhodamines, the sulfide and selene-substituted rhodamine (SR and SeR) show close spectral range as the ordinary rhodamine. The absorption and fluorescence emission are mostly less than 600 nm (Φ < 0.001) [47]. Due to the heavy atom effect of Te, the luminescence intensity of antimonyl rhodamine (2-Me-TeR) is relatively weak (Φ < 0.001).
In its oxidized product, antimonyl rhodamine (2-Me-TeOR), the heavy atom effect was weakened through the binding of an oxygen atom, giving strong fluorescence at 690 nm (Φ = 0.18) (Table 1). Recently, SO2R was synthesized by replacing the oxygen atom in rhodamine system with sulfone structure [24]. Because of the strong σ*-π* interaction between the vacant σ* orbital from the sulfur atom of and the π* system of the butadiene part, the LUMO energy level of the fluorophore got significantly lowered, thus resulting bathochromic shifted in spectra. It is worth noting that compared with SiR, SO2R exhibits a larger spectral redshift. The absorption of SO2R can reach about 700 nm, and the fluorescence emission wavelength is over 740 nm, well meets the requirements of NIR fluorescence imaging of biological samples.
|
|
Table 1 The optical properties of substituted rhodamines. |
{kind=link}
{kind=link}
3.2. Equilibrium of the spirolactone and zwitterion
In the design of fluorescent probes, fluorophores with controllable fluorescence are particular favored for the fluorescent imaging of targets with high sensitivity and selectivity. One of the major reasons for the extensive application of xanthene dyes in fluorescent imaging is the feasible regulation of fluorescence by spirocyclization for rhodamines with spiro ring. In rhodamine fluorophores with spiro rings, the closed derivative (spirolactone form) is nonfluorescent, whereas the opened ones (zwitterion form) are highly fluorescent. Thus, the stability of spiro ring plays a decisive role in the manipulation of fluorescence. The substituted rhodamine with tuned electronic properties has largely perturbed the stability of the spiro rings, thus affected the switching of fluorescence.
The introduction of Si atom into the bridging region of rhodamines can efficiently shifted the equilibrium into their spirolactone forms, compared with their oxygen analogues. Sirhodamine which favors spirolactone form has resulted fluorescent imaging with low background signal and high signal-to-noise ratio [15, 16, 48-50]. We have also proved that the Si atom can efficiently lock the spiro ring with bathochromic-shifted spectra at NIR region, even at acidic environment with pH as low as 5.0 [51]. The unusual but excellent stability of the spirolactam in the developed fluorescent probe SiRB-NO has minimized the background fluorescence under acidic condition, offering excellent sensitivity and signal/noise ratios in acidic lysosomes (Fig. 3). Moreover, the ring-opened, highly fluorescent Si-rhodamine structure is also quite stable in acidic condition, giving persistent fluorescence for long-term imaging experiments. Benefiting from the excellent stability and NIR spectra of Si-rhodamine, the developed lysosome-targeting probe has been successfully applied in monitoring both exogenous and endogenous nitric oxide (NO) in acidic lysosome at subcellular level in vivo.

|
Download:
|
| Fig. 3. luorescent imaging of lysosome in HepG2 with SiRB-NO (a-d) and SiR-B (e– h). (a–d) Reproduced with permission [51]. Copyright 2016, Wiley. (e–h) Reproduced with permission [52]. Copyright 2015, Royal Society of Chemistry. | |
{kind=link}
The excellent stability of SiR with either spirolactone or zwitterion structures has offered an ideal skeleton for the fluorescent imaging of pH in living organisms. We have also successfully incorporated a cyclic boronate structure into SiR and achieved fluorophore SiR-B with reversible transformation between boronate and boronic acid at different pH [52]. Both SiR-B and its rhodamine analogous R-B shared similar reversible H+- triggered ring opening process with the switching of fluorescence. However, compared with R-B, the strong electrophilicity of the SiR has stabilized the spirocyclic structures. The exaggerated stability in SiR-B structure has resulted in significant shift of pKcycl from basic region in R-B (9.6) into acidic region in SiR-B (6.2). The probe proteins [15]. SiR-B has offered general suitability of specific labelling of acidic lysosomes and tracking pH changes in living cells as a NIR fluorophore (Fig. 3).
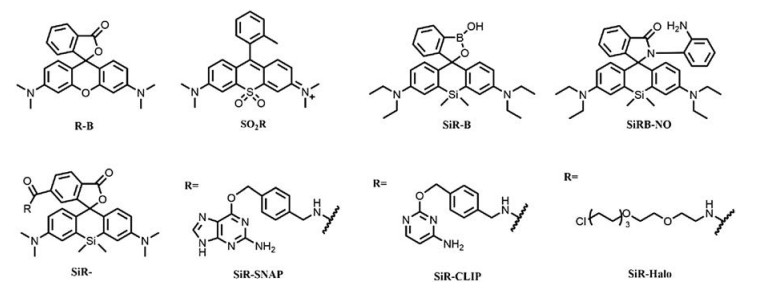
The Si-rhodamine fluorophores with spirolactone structures have also been successfully applied in the imaging of different organelles in living cells through labelling of fusion proteins of SNAP-, CLIP- and Halo-tags [15]. The developed fluorophores have performed excellent NIR fluorescent imaging results with high efficiency and selectivity, even without washing process [53]. SiRSNAP, SiR-CLIP and SiR-Halo can react efficiently with respective targets, yielding ring-opened fluorophores with strong NIR fluorescence. Low background of SiR derivatives confirmed their existence as the non-fluorescent spirolactone in the absence of targets (Fig. 4). This is likely ascribed to the formation of aggregates or unspecific binding to hydrophobic structures of the developed SiR probes in vivo. This hypothesis has been further confirmed in the fluorescent imaging of cytoskeleton in live cell (Fig. 5) [48]. The aggregation of SiR derivatives kept them as spirolactone form without fluorescence, while the interaction with polar protein surfaces have switched them into zwitterion form and yielding strong fluorescence. The fine structure of centrosomal microtubules and spatial organization of actin in the axon of cultured rat neurons have been revealed through STED imaging with developed SiR fluorophores. SiR has also been applied as a DNA stain for the imaging of living cells by STED [53].

|
Download:
|
| Fig. 4. Three-color confocal fluorescence microscopy of the tagged. Copied with permission [15]. Copyright 2013, Nature. | |
{kind=link}

|
Download:
|
| Fig. 5. Structures of some mentioned rhodamines. | |
{kind=link}
Many excellent works by other groups related to the enhanced stability of spiro-rings in Si-rhodamines are available, however, for the lack of space, we will not go to very detail in this review.
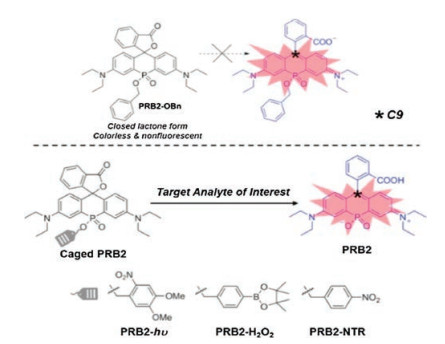
Si is not the only magic element. We have recently introduced functionalized phosphorus into the bridge region of rhodamine [21]. Other than an excellent spectral tuner as we discussed above, the phosphorus has also boosted the solubility and enhanced the photostability of the fluorophores. Moreover, after caging of the organic phosphinates at the bridge region, the stability of the spiro ring has been significantly enhanced, which locks the spironolactone in a non-fluorescent form regardless of dielectric constant. Removal of the cage resulted opening of spironolactone, accompanied with intense fluorescence. The controllable switching of fluorescence in P-rhodamine has offered a unique fluorogenic mechanism for the design of probes. We have confirmed that the functional group at the organic phosphinates in the bridge region can consequently affect the closed-open equilibrium of spirolactone in rhodamine. The positive charge of the carbon atom at the 9- position, C9, can be a straight and quantitative analyzer for the stability of spirolactone. In the example we provided, after the addition of the cage groups onto PRB2 (Fig. 6), the electrophilicity of the C9 (described by the Hirshfeld charge) has profoundly increased. Thus, the controlling in the stability of spirolactone can be efficiently used in the manipulation of fluorescence, which is feasible for the modular development of NIR probes. Moreover, phosphorus can accept different hybrid orbitals, giving large groups of organophosphorus analogues, further enriched the selection of fluorescent skeleton. NIR fluorescent probes for the sensitive imaging in vivo with the stimulation of light, H2O2 and enzyme have achieved through the modulation design of PRs.

|
Download:
|
| Fig. 6. The proposed bridge-caging strategy for the modular development of NIR fluorescent probes. Copied with permission [21]. Copyright 2018, Wiley | |
{kind=link}
For other substituted elements, disturbing at the electronic structure of xanthenes will definitely affect the stability of their sipro ring. In CR, it is likely that the spirolactone structure is more stable than the corresponding mother rhodamine, as CR is more inert with pH and dielectric constant [27]. For SO2R, the high stability of the spiro ring structure has locked it as the nonconjugated spirolactone form without color and fluorescence, regardless of pH of the environment. However, in S, Se, B, Ge and Sn substituted rhodamines, the corresponding spirolactone fluorophores are still missing. The detailed analysis of stability of spiro ring structures in substituted rhodamine, especially in SiR and PR as we discussed here, can shed some light on understanding in the controlling of fluorescence in rhodamines.
3.3. PeT efficiencyPhotoinduced electron transfer (PeT) is one of the most widely used strategies for fluorescent controlling with high efficiency. During the PeT process, fluorescence from the fluorophore can get quenched through electron transfer from the electron donor to the acceptor and vice versa [12]. Unfortunately, PeT mechanism seems unsuitable for NIR fluorophores, due to the small singlet excitation energy resulted from their long excitation wavelength [54]. However, compared to its mother rhodamine structure, SiR has bathochromic shifted spectra with inherited outstanding optical properties including high PeT efficiency. Due to the σ*-π* conjugation, replacement of oxygen by silicon has decreased both HOMO and LUMO energy level simultaneously, with a further decrease in the LUMO energy level. which are favorable for regulation of fluorescence through PeT processes [55].
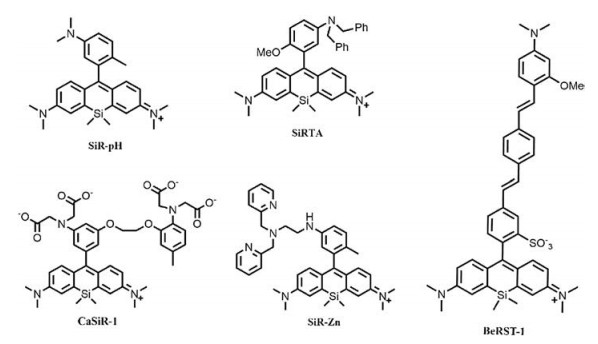
Pioneered work by Nagano and coworkers have successfully achieved PeT triggered probes in the fluorescent imaging of pH [12], Zn2+ [12], Ca2+ [14], pH sensitive SiR, SiR-Zn and CaSiR-1 (Fig. 7). In the pH sensitive SiR, the protonation and deprotonation of aniline structures on the benzene moiety were used to switch fluorescence through PeT process. Greater than 100-fold activation of the fluorophore were observed in the protonated form. SiR-Zn contains a chelating moiety, N, N-bis(2-pyridylmethyl)ethylenediamine that specific bind to Zn2+ and release fluorescence of the probe through regulation of PeT process. They have further confirmed the application of this fluorophore as an efficient turnon probe for the imaging of Zn2+ in living cells. A Ca2+ probe, CaSiR- 1 was also developed by the same group through combination of SiR and a well-know Ca2+ chelator, 1, 2-bis(o-aminophenoxy)- ethane-N, N, N', N'-tetraacetic acid moiety. Multicolor imaging for the dynamics of Ca2+ in neuronal system have proved that the combination of SiR and PeT are of great potential for NIR fluorescent imaging in biosystem.

|
Download:
|
| Fig. 7. Structures of mentioned Si-substituted rhodamines. | |
{kind=link}
Based on the aromatic tertiary amine functional group, a class of SiR probes, SiRTA have been recently reported by Guo and coworkers for fast response and high selectivity for peroxynitrite in NIR region [56]. With the existence of ONOO-, specific reaction between aromatic tertiary amine and ONOO- have reduced the electron transfer ability of benzene moiety and resulted bright fluorescence. This rapid, stable and efficient PeT process were achieved in vivo for the visualization of endogenous ONOO- during ischemia-reperfusion injury in endothelial cells, development of experimental diabetes in pancreatic β-cells and the development of diabetic nephropathy in diabetic rats (Fig. 8) [56].

|
Download:
|
| Fig. 8. (A) Imaging of ONOO- in the peritoneal cavity of rats. (B) Representative confocal images of kidney slices of rats. Copied with permission [56]. Copyright 2018, Royal Society of Chemistry. | |
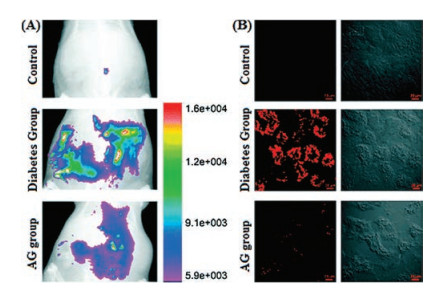{kind=link}
The PeT triggered direct optical voltage imaging was also reported by a SiR named BeRST 1 (Berkeley red sensor of transmembrane potential 1) [18]. As a small molecule probe, the developed fluorophore rapidly yields bright fluorescence with high photostability and extreme voltage-sensitivity on membrane without any genetic manipulation. Thus, the developed method can be potentially applied to all cell types for imaging of fast voltage changes with tunable color. It is worth noticing that rather than carboxyl in typical SiR, sulfonate group was applied for limiting spirocyclization of the fluorophore BeRST to nonfluorescent state in low dielectric environment such as cell membrane.
The major reason for the unusual efficient PeT process in NIR fluorophore SiR is owing to its low-lying LUMO level.We specular that PRwith evenlower LUMOlevel than SiRmight alsobe an excellentPeT triggered NIR framework. Our group is working on the develop of PR probes that switchedbyPeTmechanism andgot someexciting results, we will report this part of work in detail in near future.
3.4. Two photon absorptionTwo-photon absorption (TPA) is the simultaneous absorption of two photons for the electronic excitation of molecule [48, 49]. As an important quality for representing TPA efficiency, TPA cross section σ is the ability to arise excited states with photons of half of the nominal excitation energy. Owing to the deep penetration of long wavelength irradiation into tissues, TPA is quite attractive in the fluorescent bioimaging [57, 58]. Some pioneer works have proved that Si-rhodamine can be an excellent fluorescent scaffold for the development of two-photon fluorescent probes with decent TPA cross section σ [59, 60].
In summary, the substitution of bridging oxygen atom by other main group elements in rhodamine has not only rejuvenated this series of ancient dyes and enriched the related chemistry, but also initiated a branch of area with large and novel group of rhodamines, whose versatile structures and unique properties are highly potential in bioimaging. During the preparation of this review, a review article, which focused on different aspects and mainly summarized trend in the development of substituted rhodamine fluorophores are available [61]. Considering the excellent optical properties of substituted rhodamines, the importance of this group of fluorophores is not overstated at all. Although this group of fluorophores have already attracted increasing attentions, further research in their synthesis, properties and development of related probes are still highly desired. We believe that deep understanding in the current progress of substituted rhodamines will surely help us unveil the distinct structures and properties of fluorophores in this area.
AcknowledgmentsThe authors are grateful for the financial support from the National Natural Science Foundation of China (Nos. 21705049, 21827814), the Thousand Talent Program for Young Scholar, Shanghai Pujiang Program (No. 17PJ1402000), the Innovation Program of Shanghai Municipal Education Commission (No. 20170107005E00020) and the Program for Professor of Special Appointment (Eastern Scholar No. TP2017039).
| [1] |
S.W. Hell, S.J. Sahl, M. Bates, et al., J. Phys. D Appl. Phys. 48 (2015) 1-35. |
| [2] |
A.M. Sydor, K.J. Czymmek, E.M. Puchner, et al., Trends Cell Biol. 25 (2015) 730-748. DOI:10.1016/j.tcb.2015.10.004 |
| [3] |
S. Ma, L. Li, M. She, et al., Chin. Chem. Lett. 28 (2017) 2014-2018. DOI:10.1016/j.cclet.2017.09.027 |
| [4] |
H.R. Cheng, Y. Qian, Chin. Chem. Lett. 27 (2016) 879-886. DOI:10.1016/j.cclet.2016.01.039 |
| [5] |
X.M. Li, R.R. Zhao, Y. Yang, et al., Chin. Chem. Lett. 28 (2017) 1258-1261. DOI:10.1016/j.cclet.2016.12.029 |
| [6] |
Y. Xia, X. Liu, D. Wang, et al., Chin. Chem. Lett. 29 (2018) 1517-1520. DOI:10.1016/j.cclet.2018.01.054 |
| [7] |
X.M. Li, R.R. Zhao, Y.L. Wei, et al., Chin. Chem. Lett. 27 (2016) 813-816. DOI:10.1016/j.cclet.2016.04.001 |
| [8] |
X. Luo, L. Qian, Y. Xiao, et al., Nat. Commun 10 (2019) 258. DOI:10.1038/s41467-018-08241-3 |
| [9] |
H. He, Z. Ye, Y. Zheng, et al., Chem. Commun. (Camb.) 54 (2018) 2842-2845. DOI:10.1039/C7CC08886H |
| [10] |
H. He, T. He, Z. Zhang, et al., Chin. Chem. Lett. 29 (2018) 1497-1499. DOI:10.1016/j.cclet.2018.08.019 |
| [11] |
M. Fu, Y. Xiao, X. Qian, et al., Chem. Commun (2008) 1780-1782. |
| [12] |
Y. Koide, Y. Urano, K. Hanaoka, et al., ACS Chem. Biol. 6 (2011) 600-608. DOI:10.1021/cb1002416 |
| [13] |
Y. Koide, Y. Urano, K. Hanaoka, et al., J. Am. Chem. Soc. 133 (2011) 5680-5682. DOI:10.1021/ja111470n |
| [14] |
T. Egawa, K. Hanaoka, Y. Koide, et al., J. Am. Chem. Soc. 133 (2011) 14157-14159. DOI:10.1021/ja205809h |
| [15] |
G. Lukinavicius, K. Umezawa, N. Olivier, et al., Nat. Chem. 5 (2013) 1329-139. |
| [16] |
R.S. Erdmann, H. Takakura, A.D. Thompson, et al., Angew. Chem. Int. Ed. 53 (2014) 10242-10246. DOI:10.1002/anie.201403349 |
| [17] |
P. Shieh, M.S. Siegrist, A.J. Cullen, et al., Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 5456-5461. DOI:10.1073/pnas.1322727111 |
| [18] |
Y.L. Huang, A.S. Walker, E.W. Miller, J. Am. Chem. Soc. 137 (2015) 10767-10776. DOI:10.1021/jacs.5b06644 |
| [19] |
X. Chai, X. Cui, B. Wang, et al., Chem.-Eur. J. 21 (2015) 16754-16758. DOI:10.1002/chem.201502921 |
| [20] |
X. Zhou, R. Lai, J.R. Beck, et al., Chem. Commun. (Camb.) 52 (2016) 12290-12293. DOI:10.1039/C6CC05717A |
| [21] |
X. Chai, J. Xiao, M. Li, et al., Chem.-Eur. J. 24 (2018) 14506-14512. DOI:10.1002/chem.201802875 |
| [22] |
M. Grzybowski, M. Taki, K. Senda, et al., Angew. Chem. Int. Ed. 57 (2018) 10137-10141. DOI:10.1002/anie.201804731 |
| [23] |
X. Zhou, L. Lesiak, R. Lai, et al., Angew. Chem. Int. Ed. 56 (2017) 4197-4200. DOI:10.1002/anie.201612628 |
| [24] |
J. Liu, Y.Q. Sun, H. Zhang, et al., Acs Appl. Mater. Inter. 8 (2016) 22953-22962. DOI:10.1021/acsami.6b08338 |
| [25] |
K. Kolmakov, V.N. Belov, C.A. Wurm, et al., Eur. J. Org. Chem. 2010 (2010) 3593-3610. DOI:10.1002/ejoc.201000343 |
| [26] |
Arden-Jacob J., J. Frantzeskos, N.U. Kemnitzer, et al., Spectrochim. Acta A. 57 (2001) 2271-2283. DOI:10.1016/S1386-1425(01)00476-0 |
| [27] |
J.B. Grimm, A.J. Sung, W.R. Legant, et al., ACS Chem. Biol. 8 (2013) 1303-1310. DOI:10.1021/cb4000822 |
| [28] |
J.B. Grimm, T.D. Gruber, G. Ortiz, et al., Bioconj. Chem. 27 (2016) 474-480. DOI:10.1021/acs.bioconjchem.5b00566 |
| [29] |
G. Tombline, D.J. Donnelly, J.J. Holt, et al., Biochemistry 45 (2006) 8034-8047. DOI:10.1021/bi0603470 |
| [30] |
B. Calitree, D.J. Donnelly, J.J. Holt, et al., Organometallics 26 (2007) 6248-6257. DOI:10.1021/om700846m |
| [31] |
P. Shieh, V.T. Dien, B.J. Beahm, et al., J. Am. Chem. Soc. 137 (2015) 7145-7151. DOI:10.1021/jacs.5b02383 |
| [32] |
L.D. Lavis, Biochemistry 56 (2017) 5165-5170. DOI:10.1021/acs.biochem.7b00529 |
| [33] |
L.D. Lavis, Annu. Rev. Biochem. 86 (2017) 825-843. DOI:10.1146/annurev-biochem-061516-044839 |
| [34] |
Y. Kushida, T. Nagano, K. Hanaoka, Analyst 140 (2015) 685-695. DOI:10.1039/C4AN01172D |
| [35] |
T. Ikeno, T. Nagano, K. Hanaoka, Chem.-Asia. J 12 (2017) 1435-1446. DOI:10.1002/asia.201700385 |
| [36] |
F. Deng, Z. Xu, Chin. Chem. Lett. (2018), doi: http://dx.doi.org/10.1016/j.cclet.2018.12.012.
|
| [37] |
M. Ceresole, Ger. Pat. 44002, Nov 13, 1887.
|
| [38] |
M. Ceresole, U.S. Pat. 377349, Jan 31, 1888.
|
| [39] |
S.J. Dwight, S. Levin, Org. Lett. 18 (2016) 5316-5319. DOI:10.1021/acs.orglett.6b02635 |
| [40] |
J.B. Grimm, L.D. Lavis, Org. Lett. 13 (2011) 6354-6357. DOI:10.1021/ol202618t |
| [41] |
Y. Koide, Y. Urano, K. Hanaoka, et al., J. Am. Chem. Soc. 134 (2012) 5029-5031. DOI:10.1021/ja210375e |
| [42] |
J.L. Bachman, P.R. Escamilla, A.J. Boley, et al., Org. Lett. 21 (2019) 206-209. DOI:10.1021/acs.orglett.8b03661 |
| [43] |
B. Wang, X. Chai, W. Zhu, etal., Chem.Commun.(Camb.) 50 (2014) 14374-14377. DOI:10.1039/C4CC06178K |
| [44] |
J.B. Grimm, T.A. Brown, A.N. Tkachuk, et al., ACS Cent. Sci. 3 (2017) 975-985. DOI:10.1021/acscentsci.7b00247 |
| [45] |
C. Fischer, C. Sparr, Angew. Chem. Int. Ed. 57 (2018) 2436-2440. DOI:10.1002/anie.201711296 |
| [46] |
J.B. Grimm, B.P. English, J.J. Chen, et al., Nat. Methods 12 (2015) 244-250. DOI:10.1038/nmeth.3256 |
| [47] |
Y. Koide, M. Kawaguchi, Y. Urano, et al., Chem. Commun. (Camb.) 48 (2012) 3091-3093. DOI:10.1039/c2cc18011a |
| [48] |
G. Lukinavicius, L. Reymond, D 9'Este E., et al., Nat. Methods 11 (2014) 731-733. DOI:10.1038/nmeth.2972 |
| [49] |
J.B. Grimm, T. Klein, B.G. Kopek, et al., Angew. Chem. Int. Ed. 55 (2016) 1723-1727. DOI:10.1002/anie.201509649 |
| [50] |
G. Lukinavicius, L. Reymond, K. Umezawa, et al., J. Am. Chem. Soc. 138 (2016) 99365-9368. |
| [51] |
B. Wang, S. Yu, X. Chai, et al., Chem.-Eur. J. 22 (2016) 5649-5656. DOI:10.1002/chem.201505054 |
| [52] |
W. Zhu, X. Chai, B. Wang, et al., Chem. Commun. (Camb.) 51 (2015) 9608-9611. DOI:10.1039/C5CC02496J |
| [53] |
G. Lukinavicius, C. Blaukopf, E. Pershagen, et al., Nat. Commun 6 (2015) 8497. DOI:10.1038/ncomms9497 |
| [54] |
K. Kiyose, S. Aizawa, E. Sasaki, et al., Chem.-Eur. J. 15 (2009) 9191-9200. DOI:10.1002/chem.200900035 |
| [55] |
Y.Q. Sun, J. Liu, X. Lv, et al., Angew. Chem. Int. Ed. 51 (2012) 7634-7636. DOI:10.1002/anie.201202264 |
| [56] |
J. Miao, Y. Huo, H. Shi, et al., J. Mater. Chem. B Mater. Biol. Med. 6 (2018) 4466-4473. DOI:10.1039/C8TB00987B |
| [57] |
M. Albota, D. Beljonne, J.L. Brédas, et al., Science 281 (1998) 1653-1656. DOI:10.1126/science.281.5383.1653 |
| [58] |
F. Terenziani, C. Katan, E. Badaeva, et al., Adv. Mater. 20 (2008) 4641-4678. DOI:10.1002/adma.200800402 |
| [59] |
Z. Mao, H. Jiang, X. Song, et al., Anal. Chem. 89 (2017) 9620-9624. DOI:10.1021/acs.analchem.7b02697 |
| [60] |
H. Zhang, J. Liu, L. Wang, et al., Biomaterials 158 (2018) 10-22. DOI:10.1016/j.biomaterials.2017.12.013 |
| [61] |
L. Wang, W. Du, Z. Hu, et al., Angew. Chem. Int. Ed 58 (2019) 2-20. DOI:10.1002/anie.201813331 |