 2019, Vol. 30
2019, Vol. 30 


b University of Chinese Academy of Sciences, Beijing 100049, China

Fei Deng received his B.S. degree from Central China Normal University in 2013. He is currently a Ph.D. candidate in Dalian Institute of Chemical Physics, CAS, under the supervision of Prof. Zhaochao Xu. His research is focusing on the structure-fluorescence relationship of rhodamine dyes;

Zhaochao Xu was born in Qingdao, China, in 1979. He received his PhD in 2006 from Dalian University of Technology under the supervision of Prof. Xuhong Qian. Subsequently, he joined Prof. Juyoung Yoon's group at Ewha Womans University as a postdoctoral researcher. Since October 2008, he was a Herchel Smith Research Fellow at University of Cambridge in Prof. David R. Spring's group. In 2011, he moved to Dalian Institute of Chemical Physics, CAS, where he is currently a Professor. His research is focusing on new fluorophore design and structure-fluorescence relationship, protein labelling and sensing, the development of fluorescent probes for the selective recognition and fluorescent imaging of biologically important species, and super-resolution fluorescent imaging.
Fluorescence microscopy is an essential tool for visualizing biological processes in living cells [1-6]. The key point of this strategy is to select a proper fluorophore [7-10]. Compared with fluorescent proteins and quantum dots, organic dyes are attracting much more attention in recent 20 years, ascribed to their advantages of mall size, easy of chemical modification, good brightness and photostability, and emissions spanning the entire color spectrum [11]. Particularly, the single-molecule imaging and super-resolution imaging have been driving the development of new fluorophores with super brightness and photostability [12].
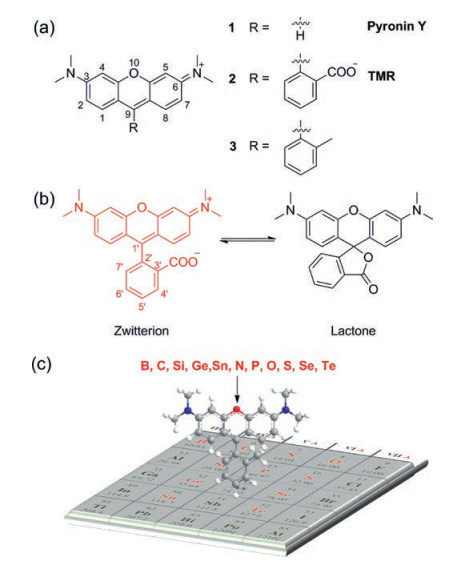
Rhodamines, a fluorophore with a history over a century, are the most popular dyes used in fluorescence imaging due to their stability, brightness and water solubility [13]. A typical structure of rhodamine is showed in Fig. 1. Although pyronine and rhodamine share the same chromophore xanthene (Fig. 1a, for example, Pyronin Y vs. tetramethylrhodamine (TMR)), rhodamine has higher brightness and stability, and is more suitable for biological application than pyronin. The carbon atom at 9-position of xanthene moiety was stabilized by the phenyl ring in rhodamine, where the one in pyronin Y was much more reactive to limit the applications of pyronine. Another feature of rhodamine is the equilibrium between the ring-opened fluorescent zwitterionic form and the ring-closed non-fluorescent lactone form (Fig. 1b). This equilibrium has been widely used to design fluorogenic chemosensors [14]. To avoid the formation of non-fluorescent lactone form, the common strategy is to introduce methyl or methoxyl groups at C-3' and C-7' in rhodamines [15, 16]. However, the absorption and emission of rhodamines within the range of 500–600 nm limit their applications in multicolour imaging and in vivo imaging [17]. These scenarios necessitate the development of near-infrared (NIR) rhodamine fluorophores.

|
Download:
|
| Fig. 1. (a) A typical structure of pyronin and rhodamine. (b) Equilibrium of TMR between zwitterionic form and lactone form. (c) Elements used in rhodamine 10-position replacement was shown in red. | |
It is required and challenging to extend the absorption and emission wavelength of rhodamines, especially to far-red and nearinfrared (NIR) region. The general strategies to elicit the absorption and emission to NIR region include π-conjugation extension and limited-flexibility of chromophore. The drawbacks of these methods are the associated decrease in brightness and watersolubility [18-20]. Another way to shift emission into NIR region is to replace the xanthene oxygen in rhodamine by heteroatoms. This strategy has been demonstrated over half century and represented by C, N, S, Se and Te-rhodamine. Due to the limited improvement in fluorescent properties and complicated synthetic routes, these rhodamines did not get much attention, until the appearance of SiPyronin in2008, pioneeredbyQian and Xiao et al. [21]. Replacement of the oxygen in the skeleton of rhodamine with silicon produces a significant red-shift to NIR regionwhilemaintaining the brightness. According to the advantage of Si-rhodamine in bioimaging, Nagano et al. developed a series of Si-rhodamines from far-red to NIR [22, 23]. Further studies revealed the fluorogenic behavior and extremely photostability of Si-rhodamine-carboxyl, which made it ideal fluorophore for live-cell super-resolution microscopy [24, 25]. The big success of Si-rhodamine has allowed a triumphant return of oxygen replacement in rhodamine modification, like borinate, phosphinate and sulfone. Here, we review various heteroatoms replaced rhodamines (Fig. 1c) and focus on their photophysical properties in order to facilitate the modification and application of new rhodamine dyes.
2. Boron groupThe boron group is the chemical elements in group 13 of the periodic table, comprising boron (B), aluminium (Al), gallium (Ga), indium (In), thallium (Tl), and perhaps also the chemically uncharacterized nihonium (Nh). At the present time, only the element of boron was reported to replace rhodamine oxygen. The first B-pyronine JS-R was reported by Egawa et al. in 2016 (Fig. 2 and Table 1, compound 4) [26]. Incorporating a borinate moiety into a xanthene skeleton produced a significant (>60 nm) bathochromic shift compared to its parent dye pyronin Y. The molar absorption coefficient and quantum yield of JS-R were measured to be 1.3 × 105 L mol-1 cm-1 and 0.59, respectively. Next, Stains et al. synthesized the corresponding B-rhodamine RF620 (Fig. 2 and Table 1, compound 5) by insertion of 2-methyl phenyl group at the 9-position of JS-R [27]. Substitution by aromatic residues caused a slight red shift (< 10 nm) in absorption and emission. Besides, molar absorption coefficient and quantum yield of RF620 were decreased to 1.09 × 105 L mol-1 cm-1 and 0.36, respectively. Similar variation between pyronin Y and TMR were observed, that TMR displayed a deceased absorption and quantum yield compared with pronin Y.

|
Download:
|
| Fig. 2. Structures of B-rhodamines. | |
|
|
Table 1 Photophysical properties of rhodamine derivatives. |
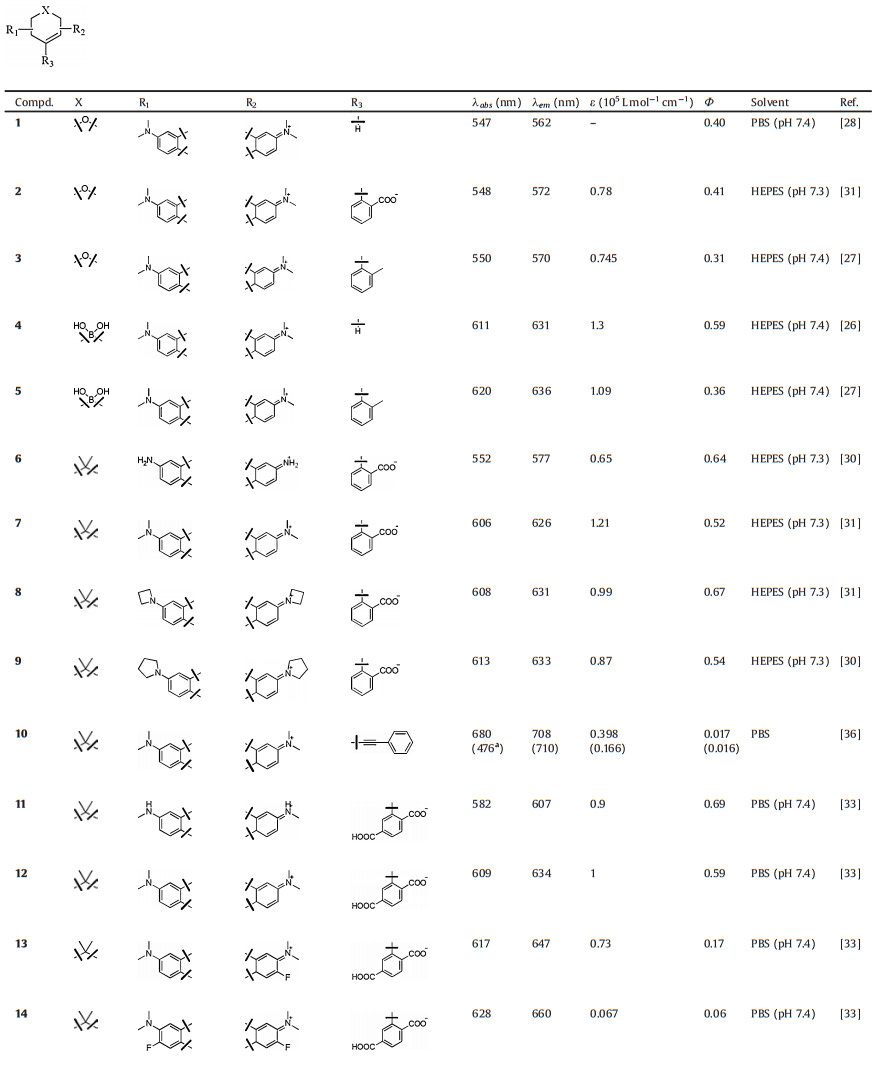


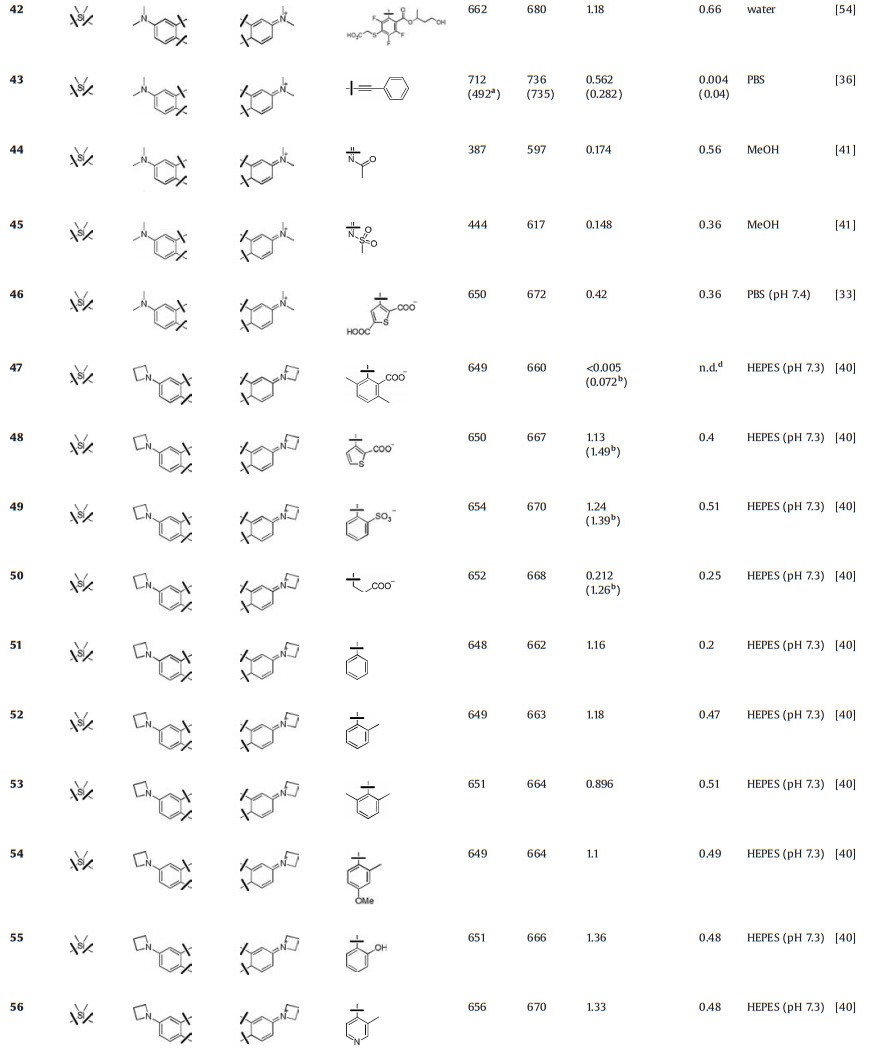

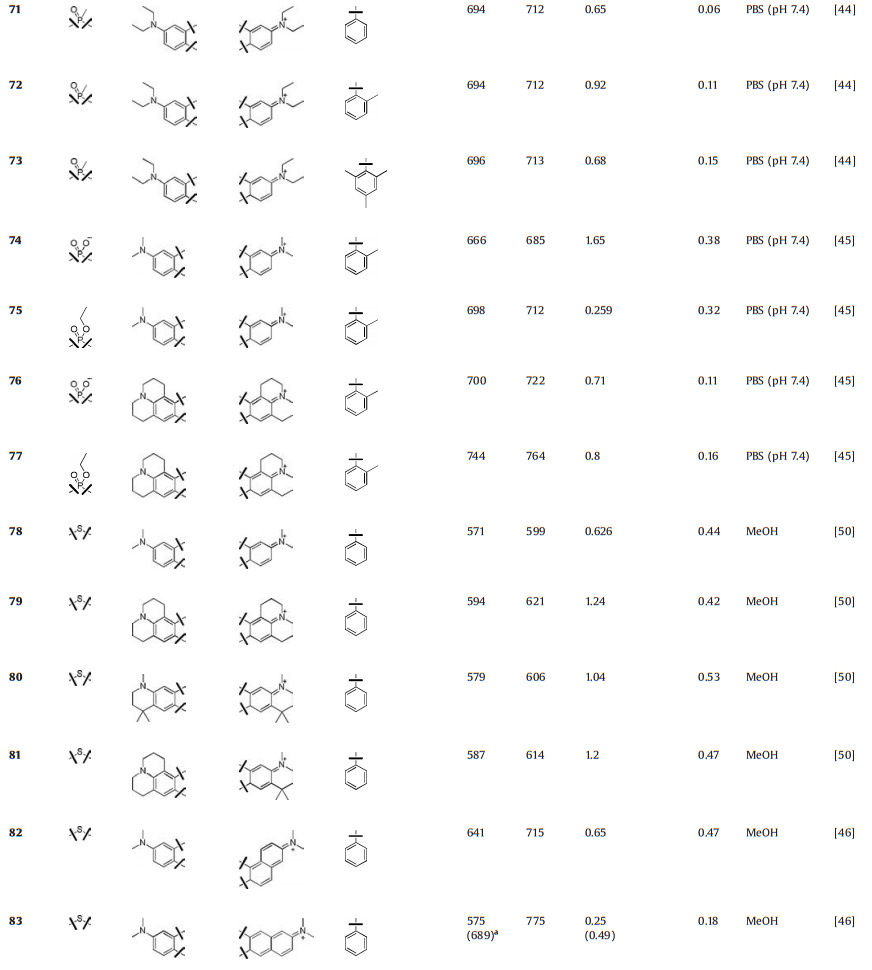
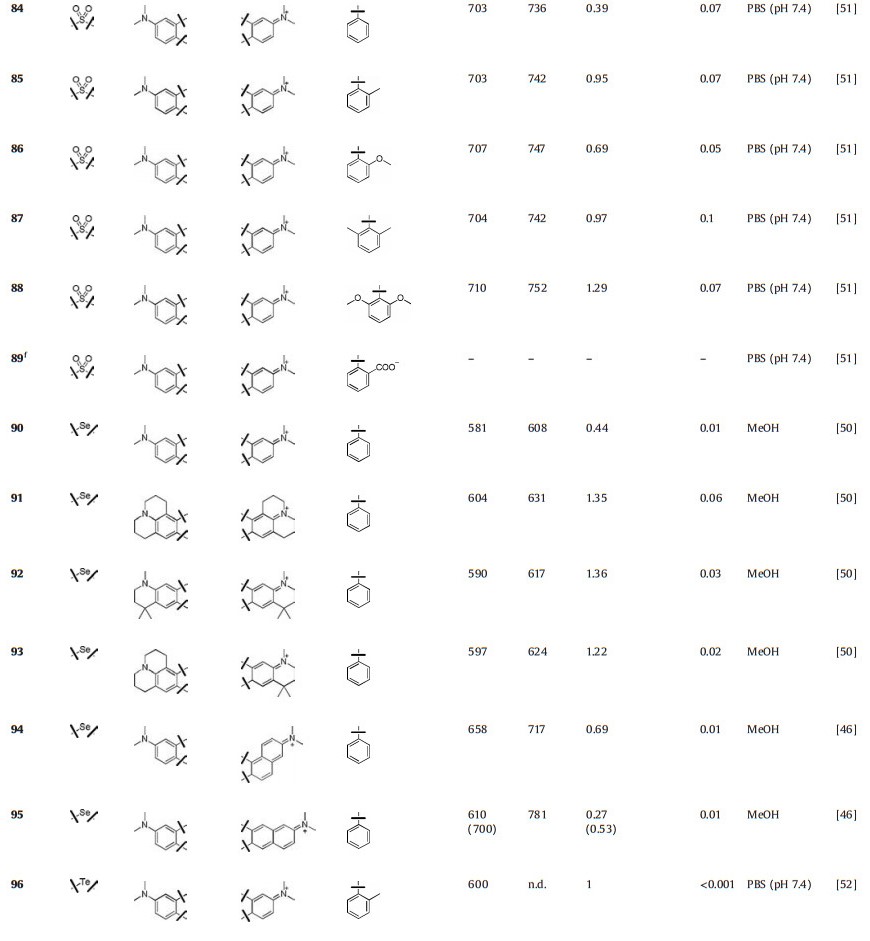
{kind=link}
{kind=link}
3. Carbon group
The carbon group, Group 14 in the p-block, contains carbon (C), silicon (Si), germanium (Ge), tin (Sn), lead (Pb) and flerovium (Fl). Except Pb and Fl, all these elements have been successfully applied in rhodamine oxygen replacement. Compared with traditional O-rhodamine, the obtained carbon-group-rhodamine fluorophores displayed significant red-shifts in fluorescence spectra. The bathochromic shift of group 14 rhodamines may be due to their lower LUMO levels. Except C-rhodamine, the existed σ*-π* conjugation in Si-, Ge- and Sn-rhodamine and the LUMO of π-system were stabilized. Besides, the conjugation became less efficient as the atomic number increase. As a consequence, the extent of red shift was C < Sn < Ge < Sn [28].
3.1. Carbon-rhodamine (C-rhodamine)Replacement of rhodamine oxygen with a quaternary carbon elicits a 50-nm bathochromic shift. These C-rhodamines were firstly synthesized by Aaron et al. in 1963 [29]. In the following half century, few attentions had been paid to the research of C-rhodamine, maybe due to the complex synthesis and low yield. Because of the high brightness and phtotostability, C-rhodamines have been successfully applied in super-resolution fluorescent imaging, which brought C-rhodamine back to the attention of dye scientists.
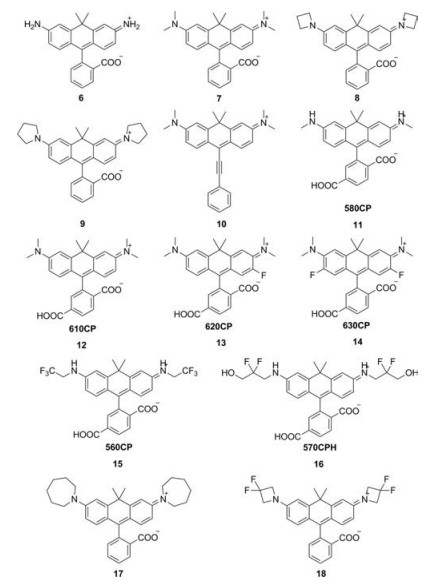
Lavis et al. reported a series of C-rhodamines by alternating the substituents on the N atoms (Fig. 3 and Table 1, compounds 6–9, 17 and 18) [30-32]. The twist of Caryl—N bond in rhodamine greatly influenced the brightness of the fluorophore. Replacing the N, N-dimethyl group in compound 7 with differently sized rings could mitigate twisted internal charge transfer (TICT) and regulate the brightness of the fluorophore. In particularly, the azetidinyl C-rhodamine (compound 8) had higher quantum yield (Φ = 0.67) compared to compound 7 (Φ = 0.52), while maintained the similar extinction coefficient (ε = 1.21 × 105 L mol-1 cm-1).

|
Download:
|
| Fig. 3. Structures of C-rhodamines. | |
{kind=link}
By introducing fluorine, Hell et al. obtained a series of C-rhodamines with maximum absorption in the range of 560–630 nm (Fig. 3 and Table 1, compounds 11–16) [33, 34]. Though the fluorination of the carbonrhodamine in tricyclic cores led to red-shifts of the absorption and emission compared to unmodified C-rhodamine, the extinction coefficients and quantum yields were reduced significantly. Taking compound 14 as an example, the extinction coefficients and quantum yields were only 6700 L mol-1 cm-1 and 0.06, whereas the unmodified compound 12 were 100, 000 L mol-1 cm-1 and 0.59, respectively. These results were totally different to O-rhodamine. Typically, the fluorination of rhodamine could lead to slight improvement in brightness [35]. However, introducing fluorine contained alkyl group into the N atoms of C-rhodamines (compounds 15 and 16) resulted in bathochromic shift while maintained the brightness compared to unmodified C-rhodamine, which was in accord with O-rhodamines.
In 2014, Klan et al. reported a NIR C-rhodamine (Fig. 3 and Table 1, compound 10) by replacing the aromatic substituents at the position C9 with phenylethynyl group [36]. This compound possessed two characteristic absorptions at 472 and 677 nm. Both of the absorption excited the maximum emission at 705 nm. The quantum yields were about 0.15 in methanol.
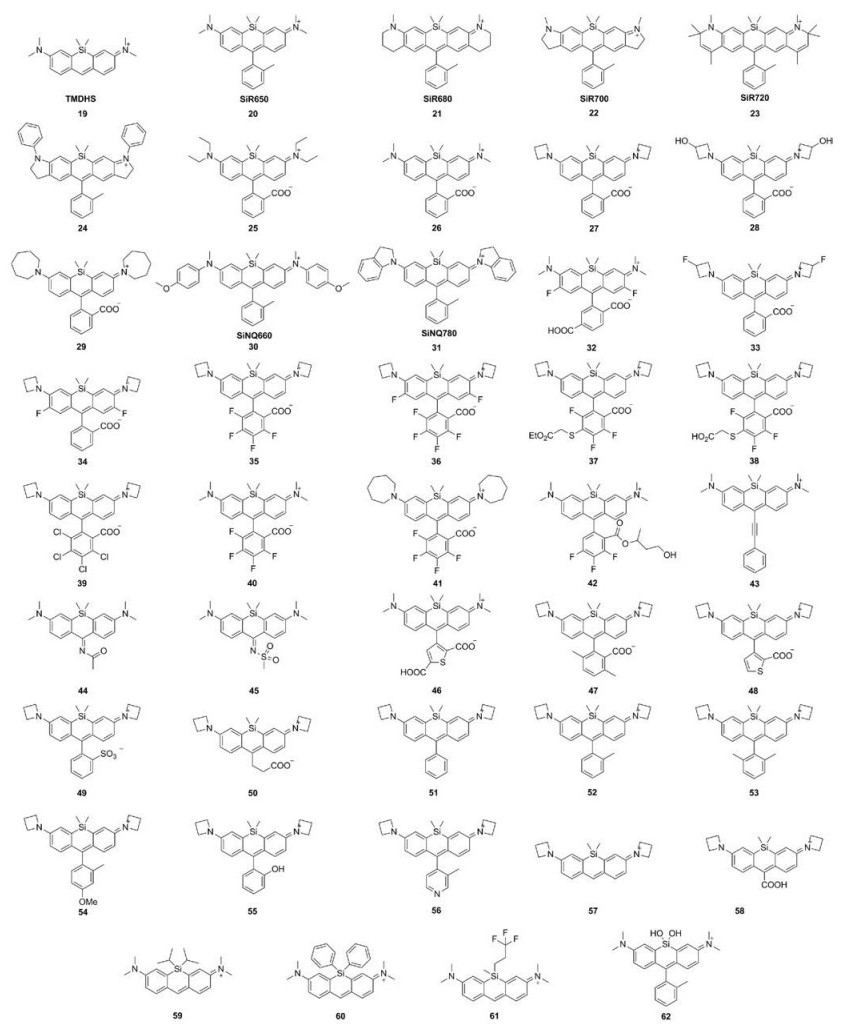
3.2. Silicon-rhodamines (Si-rhodamine)In 2008, Xiao et al. replaced the oxygen in the pyronine Y with a silicon atom to obtain TMDHS (Fig. 4 and Table 1, compound 19) [21]. The absorption and emission of TMDHS were at 641 and 659 nm, nearly 90 nm bathochromic shift compared to pyronine Y. To improve the stability, Nagano et al. inserted 2-methyl phenyl group at the 9th position of TMDHS and created Sirhodamine [28]. This compound exhibited λmax/λem = 646 nm/ 660 nm, ε = 1.1 × 105 L mol-1 cm-1 and Φ = 0.31 in PBS buffer. These data illustrated Si-rhodamine was as bright as the O-rhodamines. In order to fulfill the requirement of in vivo imaging, Nagano group further developed a series of NIR-excitable Sirhodamine (Fig. 4 and Table 1, compounds 21–24) by the expansion of the xanthene ring. These compounds show the emissions over 700 nm [37, 38]. Especially, compound 22 showed excellent tolerance to photobleaching and high quantum efficiency (Φ = 0.12) [25].

|
Download:
|
| Fig. 4. Structures of Si-rhodamines. | |
{kind=link}
Like the modification in C-rhodamines, Lavis et al. also replaced dialkylamino substituents with differently sized rings to mitigate TICT and regulate the brightness in Si-rhodamine (Fig. 4 and Table 1, compounds 25–29) [31, 34, 39, 40]. The azetidinyl Sirhodamine (compound 27) had similar absorption and emission (λmax/λem = 646 nm/664 nm) and higher quantum yield (Φ = 0.31) compared to N, N-dimethyl Si-rhodamine (compound 26). Also, depending on the free rotation of the bond between the N atom and the Si-substituted xanthene moiety, Urano et al. designed a series of near-infrared fluorescence quenchers (Fig. 4 and Table 1, compounds 30 and 31) [38]. These compounds showed absorption in NIR region (660 nm and 779 nm) and the quantum yields were almost zero.
In O-rhodamine modification, introducing halogen, especially fluorine, would improve the photostability and brightness of fluorophore. This strategy was also applied in Si-rhodamine. Lavis and Hell groups have vigorously developed various fluorine-containing Si-rhodamines (Fig. 4 and Table 1, compounds 32–42). Similar to C-rhodamine, introducing fluorine into the tricyclic cores of Si-rhodamine decreased the extinction coefficients and quantum yields sharply, albeit with nearly 30 nm red-shift in wavelengths (compounds 32, 34 and 36) [33, 40]. However, the fluorination or chlorination in the bottom phenyl group had a much smaller effect on brightness with 20–30 nm redshifts in wavelengths (compounds 35, 37–42). The fluorinated azetidine (compound 33) exhibited ~10 nm blue shift in spectral properties, a slightly higher quantum yield (Φ = 0.56) relative to compound 27, which was similar to O-rhodamines and C-rhodamines [32].
Replacing the group at the 9-position also induced fluorescence changes (Fig. 4 and Table 1, compounds 43–58). Compound 43 with a conjugated phenylethynyl group shifted the absorption and emission over 700 nm [36]. The 9-imino-10-silaxanthone compounds 44 and 45 exhibit remarkably large Stokes shifts (around 200 nm), which were related to the excitation of an electron from the HOMO to the LUMO of the chromophores [41]. These fluorophores with large Stokes shift would be useful in multicolor nanoscopy [42]. Based on the structure of azetidinyl Si-rhodamine (compound 27), Lavis et al. also changed the substituents at the 9th position. (Compounds 46–58 showed similar absorption and emission spectra (~λmax/λem = 650 nm/665 nm). The extinction coefficients of these compounds were about 1.2 × 105 L mol-1 cm-1. However, the quantum yields were greatly influenced by the substitutes. For example, compound 51 had a lower quantum yield of 0.2, while the quantum yields of compounds 52–56 were over 0.5 [33, 40]. The intramolecular rotation of phenyl ring in 51 may decrease the quantum yield.
Dimethylsilane was routinely used as heteroatom in Sirhodamine. Indeed, the different substituents on silicon atoms also affect the fluorescence properties. For example, compounds 59–61 with different Si-substitutes were developed by Zhang et al. (Fig. 4 and Table 1). These compounds displayed different bathochromic shifts and quantum yields [43]. For compound 62, the substitute was changed from silane to silanediol, and the excitation and emission were further red-shifted to 663 nm and 681 nm, respectively, with ε = 1.05 × 105 L mol-1 cm-1 and Φ = 0.43 in PBS buffer [27].
3.3. Germanium-rhodamines (Ge-rhodamine)Ge-rhodamines display further about 10 nm hypsochromic shift compared with Si-rhodamine. And the brightness is similar with that of Si-rhodamine (Fig. 5 and Table 1, compounds 63–66) [28, 34]. Taking compound 65 as an example, it displayed λmax/λem = 410 nm/471 nm, ε = 9.7 × 104 L mol-1 cm-1 and Φ = 0.43. Although the attention to Ge-rhodamine is constrained by the fact that synthetic raw materials are not readily available, the outstanding brightness and proper excitation wavelength make Ge-rhodamine a promising fluorophore in bioimaging (Fig. 5).

|
Download:
|
| Fig. 5. Structures of Ge-rhodamines. | |
{kind=link}
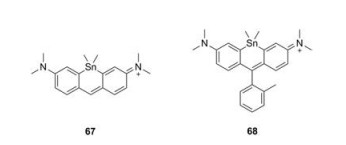
3.4. Tin-rhodamines (Sn-rhodamine)
Compared to C-, Si- and Ge-rhodamine, Sn-rhodamines were rarely reported (Fig. 6 and Table 1, compounds 67–68) [28]. Nagano group synthesized both Sn-pyronine and Sn-rhodamine and found they were really chemical-active. Compound 68 showed the maximum absorption and emission at 614 nm and 628 nm, respectively.

|
Download:
|
| Fig. 6. Structures of tin-substituted rhodamines. | |
{kind=link}
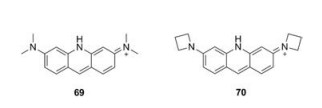
4. Nitrogen family 4.1. Nitrogen-rhodamines (N-rhodamine)
Replacement of the oxygen by a nitrogen atom on the pyronin framework produced acridine orange (69), which have been widely used as a nucleic acid-selective dye over half a century. When bound to DNA, acridine orange displayed a similar emission with that of fluorescein. When bound to RNA, its excitation and emission were shifted to 460 nm and 650 nm, respectively. Lavis et al. replaced the N, N-dimethylamino substituents in acridine orange with four-membered azetidine rings. Compound 70 showed an improved quantum yield from 0.21 to 0.52 (Fig. 7 and Table 1, compounds 69–70) [31].

|
Download:
|
| Fig. 7. Structures of N-rhodamines. | |
{kind=link}
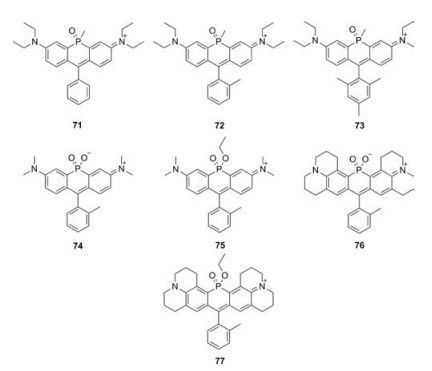
4.2. Phosphorus-rhodamines (P-rhodamine)
Besides nitrogen, phosphorus was also used to replace rhodamine oxygen. In 2015, Wang et al. reported a series of Prhodamines (Fig. 8 and Table 1, compounds 71–73) [44]. Due to the electron-withdrawing properties of the phosphorus moiety, these P-rhodamines elicit 140 nm bathochromic shifts relative to O-rhodamine. These compounds displayed similar absorption and emission spectra (λmax/λem = 694 nm/711 nm). Due to the restricted intramolecular rotation, the quantum yields of 71-73, which have increasing number of methyl substituents in phenyl group, improved from 0.06 to 0.15. Stains et al. used phosphinate functional group as the bridge and created P-rhodamines 74–77 (Fig. 8 and Table 1). Compound 74 exhibited excitation and emission maxima at 666 nm and 685 nm, respectively. The molar extinction coefficients and quantum yields were 1.65 × 105 L mol-1 cm-1 and Φ = 0.38, respectively. Moreover, its ethyl ester counterpart compound 75 showed further 35 nm bathochromic shift, though the brightness decreased. By replacing the dimethylaniline in compounds 74 and 75 with julolidine substituent, the excitation and emissions in compounds 76 and 77 were further red-shifted to the rang over 700 nm [45].

|
Download:
|
| Fig. 8. Structures of P-rhodamines. | |
{kind=link}
5. Oxygen family
Due to the similar chemical characteristics in chalcogens, it was reasonable to replace the bridging oxygen atom with other chalcogens. The extent of red shift in emissions was correlate with the atom size (O < S < Se < Te) [46]. This trend was thought to be related to the resonance effect of the chalcogen atom, which narrowed the HOMO-LUMO gap [47, 48]. Besides, the molar extinction coefficients and fluorescence quantum yields decreased with the increasing size of the chalcogen atom, which could be attributed to a strong heavy-atom effect [49]. Different with oxygen, the common oxidation states in S, Se, and Te could be -2, +4 and +6. The corresponding oxide can also be applied in replacing the bridging oxygen atom.
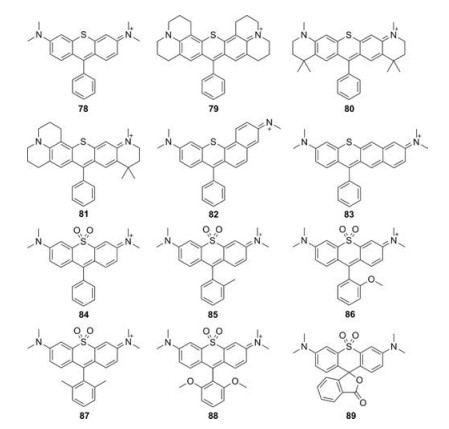
5.1. Sulfur-rhodamines (S-rhodamine)Most of S-rhodamines were firstly reported by Detty group (Fig. 9 and Table 1, compounds 78–83). Compared to O-rhodamine, S-rhodamines displayed about 20 nm red-shift in absorption and emission spectra. However, the brightness was less than half that of O-rhodamine. Taking compound 78 as an example, it exhibited λmax/λem = 571 nm/599 nm, ε = 6.26 × 104 L mol-1 cm-1 and Φ = 0.44 in methanol. These photophysical properties limited the wide applications of S-rhodamine in biological imaging [46, 49, 50].

|
Download:
|
| Fig. 9. Structures of S-rhodamines and sulfone-rhodamines. | |
{kind=link}
Guo et al. reported a series of sulfone-rhodamines in 2016 (Fig. 9 and Table 1, compounds 84–89) [51]. The sulfone group serves as the bridge to rigidify their structures and a strong electron withdrawing group. The absorption and emission of sulfone-rhodamines reached 700 nm and 730 nm, respectively. Different substituents in phenyl group influenced the stability and brightness due to the steric effects, which have been referred in P-rhodamines.
5.2. Selenium-rhodamines (Se-rhodamine)When the oxygen bridge was replaced by Selentium, the bathochromic shift in emission was further increased by 30 nm associated with sharply decreased brightness (Fig. 10 and Table 1, compounds 90–95) [46, 50]. For example, compound 90 showed λmax/λem = 581 nm/608 nm and ε = 4.4 × 104 L mol-1 cm-1, but a relatively low Φ = 0.01 in methanol. Unlike other dyes, Serhodamine had a high yield for singlet oxygen generation, which could be applied as an efficient photosensitizer [49].

|
Download:
|
| Fig. 10. Structures of Se-rhodamines. | |
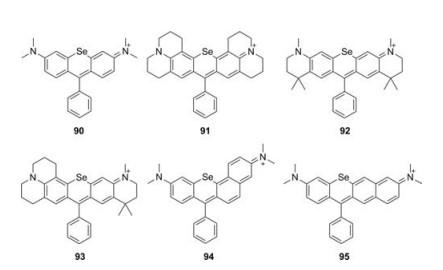{kind=link}
5.3. Tellurium-rhodamines (Te-rhodamine)
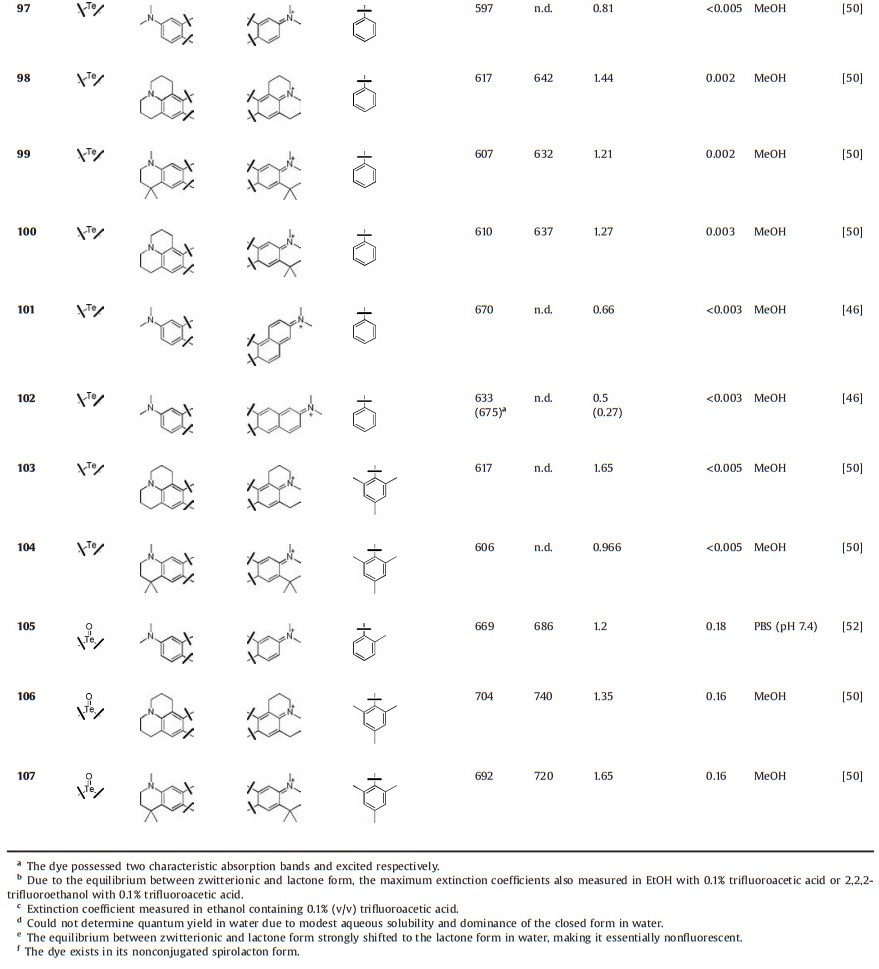
Te-rhodamines were reported with very weak fluorescence (Φ < 0.001) due to the heavy-atom effect (Fig. 11 and Table 1, compounds 96–104) [50, 52, 53]. For Te-rhodamines, Te atom could be easily oxidized by reactive oxygen species (Fig. 11 and Table 1, compounds 105–107). The corresponding telluroxide rhodamines exhibited a large red shift compared to Te-rhodamine and showed strong fluorescence. Taking compound 96 as an example, it could be oxidized to compound 105 by reactive oxygen species and exhibited maximum fluorescence emission around 686 nm with Φ = 0.18 [52]. These results indicated that the heavy-atom effect could be weakened by binding of oxygen atom.

|
Download:
|
| Fig. 11. Structures of Te-rhodamines. | |
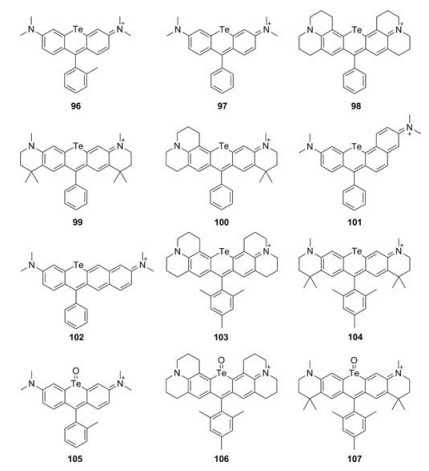{kind=link}
6. Conclusions and perspectives
Rhodamine is a type of widely used fluorophore. The bridge modification atom at 10 position enriches the color palette of rhodamines. So far, most of the possible element have been applied to build heteroatom-substituted rhodamine. Changing the functional group of the same element at 10 position seems a promising method to further extend the heteroatom-substituted rhodamines in the further. For example, sulfur-rhodamine and sulfonerhodamine share the same element at 10 position but have totally different photophysical properties. Besides, most of the researches in this field are focusing on group 14 elements, especially silicon. A number of methods have been proposed to improve the brightness, photostability and fluorogenicity of rhodamine, C-rhodamine and Si-rhodamine. Among these methods, incorporation of fourmembered azetidine rings into the fluorophore is one of the most attractive. However, these methods have rarely been applied in other element replaced rhodamines so far. We hope that this review paper can draw much more attention on the structural modification of rhodamines. A new way of thinking can be found through the comparison of fluorescence structure-activity relationships. We hope that the structure-activity relationship summarized here, as shown in Table 1, will help to achieve the goal of creating more dyes with high brightness and photostability.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation China (No. 21878286) and DICP (Nos. DMTO201603, TMSR201601).
| [1] |
Y. Kang, J.L. Fan, Q. Jin, et al., Chin. Chem. Lett. 28 (2017) 1991-1993. DOI:10.1016/j.cclet.2017.08.054 |
| [2] |
L.X. Peng, Y.X. Xu, P. Zou, Chin. Chem. Lett. 28 (2017) 1925-1928. DOI:10.1016/j.cclet.2017.09.037 |
| [3] |
P. Ning, W.J. Wang, M. Chen, Y. Feng, X.M. Meng, Chin. Chem. Lett. 28 (2017) 1943-1951. DOI:10.1016/j.cclet.2017.09.026 |
| [4] |
D. Wu, Y.Z. Shen, J.H. Chen, et al., Chin. Chem. Lett. 28 (2017) 1979-1982. DOI:10.1016/j.cclet.2017.07.004 |
| [5] |
S. Leng, Q.L. Qiao, Y. Gao, et al., Chin. Chem. Lett. 28 (2017) 1911-1915. DOI:10.1016/j.cclet.2017.03.034 |
| [6] |
F. Yang, C. Wang, L. Wang, et al., Chin. Chem. Lett. 28 (2017) 2019-2022. DOI:10.1016/j.cclet.2017.07.030 |
| [7] |
H. Zhou, Y. Xiao, X. Hong, Chin. Chem. Lett. 29 (2018) 1425-1428. DOI:10.1016/j.cclet.2018.08.009 |
| [8] |
W. Chen, Y. Pan, J. Chen, et al., Chin. Chem. Lett. 29 (2018) 1429-1435. DOI:10.1016/j.cclet.2018.08.011 |
| [9] |
X. Ma, L. Hu, X. Han, J. Yin, Chin. Chem. Lett. 29 (2018) 1489-1492. DOI:10.1016/j.cclet.2018.06.022 |
| [10] |
G. Gao, Y.W. Jiang, W. Sun, F.G. Wu, Chin. Chem. Lett. 29 (2018) 1475-1485. DOI:10.1016/j.cclet.2018.07.004 |
| [11] |
L.D. Lavis, Biochemistry 56 (2017) 5165-5170. DOI:10.1021/acs.biochem.7b00529 |
| [12] |
van de Linde S., M. Heilemann, M. Sauer, Annu. Rev. Phys. Chem. 63 (2012) 519-540. DOI:10.1146/annurev-physchem-032811-112012 |
| [13] |
L.D. Lavis, R.T. Raines, ACS Chem. Biol. 3 (2008) 142-155. DOI:10.1021/cb700248m |
| [14] |
M. Beija, C.A.M. Afonso, J.M.G. Martinho, Chem. Soc. Rev. 38 (2009) 2410-2433. DOI:10.1039/b901612k |
| [15] |
L.X. Wu, K. Burgess, J. Org. Chem. 73 (2008) 8711-8718. DOI:10.1021/jo800902j |
| [16] |
Z.H. Lei, X.R. Li, Y. Li, et al., J. Org. Chem. 80 (2015) 11538-11543. DOI:10.1021/acs.joc.5b01746 |
| [17] |
T. Ikeno, T. Nagano, K. Hanaoka, Chem.-Asian J. 12 (2017) 1435-1446. DOI:10.1002/asia.201700385 |
| [18] |
Y.Q. Sun, J. Liu, X. Lv, et al., Angew. Chem. Int. Ed. 51 (2012) 7634-7636. DOI:10.1002/anie.201202264 |
| [19] |
L. Yuan, W.Y. Lin, K.B. Zheng, L.W. He, W.M. Huang, Chem. Soc. Rev. 42 (2013) 622-661. DOI:10.1039/C2CS35313J |
| [20] |
M.T.Z. Spence, I.D. Johnson, The Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies, 11th ed., Live Technologies Corporation, Carlsbad, CA, 2010.
|
| [21] |
M.Y. Fu, Y. Xiao, X.H. Qian, D.F. Zhao, Y.F. Xu, Chem. Commun. (2008) 1780-1782. |
| [22] |
T. Egawa, K. Hanaoka, Y. Koide, et al., J. Am. Chem. Soc. 133 (2011) 14157-14159. DOI:10.1021/ja205809h |
| [23] |
Y. Koide, Y. Urano, K. Hanaoka, T. Terai, T. Nagano, J. Am. Chem. Soc. 133 (2011) 5680-5682. DOI:10.1021/ja111470n |
| [24] |
G. Lukinavicius, K. Umezawa, N. Olivier, et al., Nat. Chem. 5 (2013) 132-139. DOI:10.1038/nchem.1546 |
| [25] |
G. Lukinavicius, L. Reymond, K. Umezawa, et al., J. Am. Chem. Soc. 138 (2016) 9365-9368. DOI:10.1021/jacs.6b04782 |
| [26] |
N. Shimomura, Y. Egawa, R. Miki, T. Fujihara, Y. Ishimaru, T. Seki, Org. Biomol. Chem. 14 (2016) 10031-10036. DOI:10.1039/C6OB01695B |
| [27] |
X.Q. Zhou, L. Lesiak, R. Lai, et al., Angew. Chem. Int. Ed. 56 (2017) 4197-4200. DOI:10.1002/anie.201612628 |
| [28] |
Y. Koide, Y. Urano, K. Hanaoka, T. Terai, T. Nagano, ACS Chem. Biol. 6 (2011) 600-608. DOI:10.1021/cb1002416 |
| [29] |
C. Aaron, C.C. Barker, J. Chem. Soc. 5 (1963) 2655-2662. |
| [30] |
J.B. Grimm, A.J. Sung, W.R. Legant, et al., ACS Chem. Biol. 8 (2013) 1303-1310. DOI:10.1021/cb4000822 |
| [31] |
J.B. Grimm, B.P. English, J.J. Chen, et al., Nat. Methods 12 (2015) 244-250. DOI:10.1038/nmeth.3256 |
| [32] |
J.B. Grimm, A.K. Muthusamy, Y. Liang, et al., Nat. Methods 14 (2017) 987-994. DOI:10.1038/nmeth.4403 |
| [33] |
A.N. Butkevich, G.Y. Mitronova, S.C. Sidenstein, et al., Angew. Chem. Int. Ed. 55 (2016) 3290-3294. DOI:10.1002/anie.201511018 |
| [34] |
A.N. Butkevich, V.N. Belov, K. Kolmakov, et al., Chem.-Eur. J. 23 (2017) 12114-12119. DOI:10.1002/chem.201701216 |
| [35] |
G.Y. Mitronova, V.N. Belov, M.L. Bossi, et al., Chem.-Eur. J. 16 (2010) 4477-4488. DOI:10.1002/chem.200903272 |
| [36] |
T. Pastierik, P. Sebej, J. Medalova, P. Stacko, P. Klan, J. Org. Chem. 79 (2014) 3374-3382. DOI:10.1021/jo500140y |
| [37] |
Y. Koide, Y. Urano, K. Hanaoka, et al., J. Am. Chem. Soc. 134 (2012) 5029-5031. DOI:10.1021/ja210375e |
| [38] |
T. Myochin, K. Hanaoka, S. Iwaki, et al., J. Am. Chem. Soc. 137 (2015) 4759-4765. DOI:10.1021/jacs.5b00246 |
| [39] |
T. Wang, Q.J. Zhao, H.G. Hu, et al., Chem. Commun. 48 (2012) 8781-8783. DOI:10.1039/c2cc34159j |
| [40] |
J.B. Grimm, T.A. Brown, A.N. Tkachuk, L.D. Lavis, ACS Cent. Sci. 3 (2017) 975-985. DOI:10.1021/acscentsci.7b00247 |
| [41] |
P. Horvath, P. Sebej, T. Solomek, P. Klan, J. Org. Chem. 80 (2015) 1299-1311. DOI:10.1021/jo502213t |
| [42] |
A.N. Butkevich, G. Lukinavicius, D'Este E., S.W. Hell, J. Am. Chem. Soc. 139 (2017) 12378-12381. DOI:10.1021/jacs.7b06412 |
| [43] |
H.L. Nie, L. Qiao, W. Yang, et al., J. Mater. Chem. B 4 (2016) 4826-4831. DOI:10.1039/C6TB00938G |
| [44] |
X.Y. Chai, X.Y. Cui, B.G. Wang, et al., Chem.-Eur. J. 21 (2015) 16754-16758. DOI:10.1002/chem.201502921 |
| [45] |
X.Q. Zhou, R. Lai, J.R. Beck, H. Li, C.I. Stains, Chem. Commun. 52 (2016) 12290-12293. DOI:10.1039/C6CC05717A |
| [46] |
M.W. Kryman, T.M. McCormick, M.R. Detty, Organometallics 35 (2016) 1944-1955. DOI:10.1021/acs.organomet.6b00255 |
| [47] |
M.R. Detty, P.N. Prasad, D.J. Donnelly, et al., Bioorg. Med. Chem. 12 (2004) 2537-2544. DOI:10.1016/j.bmc.2004.03.029 |
| [48] |
B. Calitree, D.J. Donnelly, J.J. Holt, et al., Organometallics 26 (2007) 6248-6257. DOI:10.1021/om700846m |
| [49] |
T.Y. Ohulchanskyy, D.J. Donnelly, M.R. Detty, P.N. Prasad, J. Phys. Chem. B 108 (2004) 8668-8672. DOI:10.1021/jp0370674 |
| [50] |
M.W. Kryman, G.A. Schamerhorn, J.E. Hill, et al., Organometallics 33 (2014) 2628-2640. DOI:10.1021/om500346j |
| [51] |
J. Liu, Y.Q. Sung, H.X. Zhang, et al., ACS Appl. Mater. Interfaces 8 (2016) 22953-22962. DOI:10.1021/acsami.6b08338 |
| [52] |
Y. Koide, M. Kawaguchi, Y. Urano, et al., Chem. Commun. 48 (2012) 3091-3093. DOI:10.1039/c2cc18011a |
| [53] |
M.W. Kryman, G.A. Schamerhorn, K.Y. Yung, et al., Organometallics 32 (2013) 4321-4333. DOI:10.1021/om400467s |
| [54] |
K. Kolmakov, E. Hebisch, T. Wolfram, et al., Chem.-Eur. J. 21 (2015) 13344-13356. DOI:10.1002/chem.201501394 |