 2019, Vol. 30
2019, Vol. 30 


b Department of Biology, University of Waterloo, Waterloo, Ontario, N2L 3G1, Canada
Estrogens are the primary female sex hormones that play critical roles in reproductive systems and secondary sex characteristics [1, 2]. For example, estradiol (E2), the major estrogenic compound, is responsible for the regulation of estrous and menstrual cycles [3]. However, accumulation of environmental estrogens due to wastewater effluents and pharmaceuticals discharges has raised prominent concerns [4, 5]. For example, estrogens from municipal wastewater were reported to feminization of wild fish [6]. Exposure to low concentrations of environmental estrogens interferes with human endocrine systems [7].
To minimize the hazard, many methods have been developed to degrade estrogens through water treatment by using microorganism [8], ozone [9], ultrasound [10], UV and hydrogen peroxide or their combinations [11]. Photocatalysis was also reported to be capable of estrogen degradation [12]. For example, estradiol (E2) was degraded using TiO2 combined with UV light [13]. Although effective, these methods are complicated, high-cost, or need energy input.
Many inorganic nanomaterials have enzyme-like activities under ambient conditions, and they are called nanozymes [14-18]. For example, gold nanoparticles (AuNPs) have glucose oxidase activity [19, 20], iron oxide nanoparticles have peroxidase activity [21], and nanoceria has oxidase and other types of redox activities [22, 23]. With the advantages of high stability and low cost, nanozymes are attractive in various applications ranging from biosensor development [24-26] to nanomedicine [27-30]. So far, most nanozymes have focused on some model chromogenic and fluorogenic substrates [31-37], while their conversion of environmentally and biologically important substrates is much less explored [38-40]. For example, nanozymes have not yet been tested for estrogen-based substrates. Part of the difficulty is associated with the detection. Without an optical signal change, more advanced techniques have to be used for the analysis.
In the present study, we screened six common nanozymes using estrogen compounds as substrates to determine the potential catalytic activity. We found that AuNPs can catalyze E2 oxidation to E1 in a simple aqueous solution without the need of extra chemicals or energy input. Therefore, the AuNPs showed a dehydrogenase-like activity in this case.
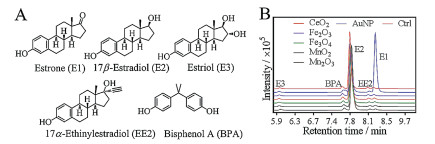
Five common estrogen compounds were tested (Fig. 1A), including three natural estrogens: estrone (E1), 17β-estradiol (E2) and estriol (E3) (named as the number of hydroxyl group contained), and two synthetic estrogens 17α-ethinylestradiol (EE2) and bisphenol A (BPA). Since E2 is the most potent estrogen, we first screened the nanomaterials with E2. AuNPs, nanoceria (CeO2), Fe3O4, Fe2O3, MnO2 and Mn2O3 nanoparticles were chosen for screening. These nanomaterials were spherical and their sizes ranged from ~5 nm to 50 nm, with nanoceria being the smallest of ~5 nm and Mn2O3 being the largest of ~50 nm [41]. These nanoparticles were respectively incubated with E2. Then after centrifugation to precipitate the nanoparticles, the supernatants were quantitatively measured by using LC-MS. To determine the retention time of each estrogen in the LC-MS spectra, a set of standards containing all these estrogens were also added to each sample before analysis, and these peaks can be observed from the control sample without any nanomaterial added (red trace, Fig. 1B). For the nanomaterial treated samples, only the AuNP added sample produced a new peak (at ~8.3 min retention time), suggesting that it could convert E2 to another compound.

|
Download:
|
| Fig. 1. (A) Chemical structures of the estrogens used in this study. BPA was also included as an environmental contaminant for analysis. (B) The LC-MS spectra of E2 only sample (Ctrl) and E2 mixed with different nanozymes for 20 min in HEPES buffer (20 mmol/L, pH 7) at room temperature. The other peaks were from the spiked standards to assign the peaks. | |
{kind=link}
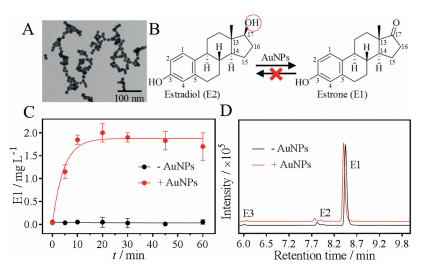
Since only the AuNPs showed activity, we focused our study on it. The AuNPs used here were ~13 nm in diameter (Fig. 2A). From the retention time of each estrogen on the control spectra by running the standard samples (Fig. 1B) [42, 43], E2 had a peak at ~7.8 min and the converted product at ~8.3 min was identified to be E1. The production of E1 was also confirmed from the mass spectrometry (MS) data (Fig. S1 in Supporting information). Other estrogens with small peaks were only from the standards (Fig. 1B). Therefore, the AuNPs catalyzed the conversion of E2 to E1 (Fig. 2B).

|
Download:
|
| Fig. 2. (A) A TEM photo of 13 nm AuNPs. (B) AuNPs catalyze oxidation of E2 to E1 and the reaction is irreversible. The red circled group of E2 is 17β-OH. (C) Kinetics of the converted E1 from E2 oxidation with or without AuNPs. (D) The LC-MS spectra of E1 with or without mixed with AuNPs for 1 h. The small peaks of E2 and E3 were from the spiked standards to assign the peaks. E2 or E1 (0.1 mmol/L) and 13 nm AuNPs (5 nmol/L) were used for the reactions in HEPES buffer (20 mmol/L, pH 7) at room temperature. | |
{kind=link}
To determine the reaction rate, we then measured the kinetics by plotting the converted E1 as a function a reaction time (red trace, Fig. 2C). The reaction was very fast, since the converted E1 reached to the maximum of ~2.0 mg/L within just 10 min, and the first-order rate constant was calculated to be 0.22 min-1. At the same time, no E1 was measured for the samples without AuNPs, confirming the high catalytic activity of AuNPs (black trace, Fig. 2C).
The 17β-OH group in E2 was converted to a keto in E1 (Fig. 2B), and this is a typical dehydrogenation reaction (a type of oxidation). This oxidation mechanism has been well studied in the AuNPs catalyzed alcohol oxidation [44]. For example, AuNPs catalyze primary alcohol oxidation to carboxylic acid and convert secondary alcohol to be ketones [45]. AuNPs can also catalyze glucose oxidation to gluconate [19, 20]. In these reactions, AuNPs are mimics of oxidases with ability of transporting electrons from reducing substrates (e.g., glucose and E2) to the oxidizing agent, oxygen. E2 also has a phenol hydroxyl group at the C3 position, and this group was retained during the reaction (Fig. 2B). This fast conversion of E2 to E1 may have significant environmental implications. Since E1 has a far weaker activity as estrogen relative to E2 [46], this study provides an effective method for E2 detoxification in aqueous by simply using AuNPs.
In the metabolic pathway, E2 oxidation to E1 is usually catalyzed by 17β-hydroxysteroid dehydrogenases (17β-HSD) [46]. Since 17β-HSD is an isoenzyme complex containing both reductases and dehydrogenases, the reaction is reversible [47], i.e., oxidation from E2 to E1 and reduction from E1 to E2. To test whether the AuNPs have such reversible activity, we then mixed E1 with the AuNPs and analyzed the products in the same method (Fig. 2D). From the LC-MS spectra, aside from the added E1 with peaks at ~8.3 min, no E2 or other compounds were identified (the small peaks were from the standards). Therefore, the AuNPs worked only as an oxidase (dehydrogenase) mimic for E2 degradation and the reaction is irreversible (Fig. 2B).
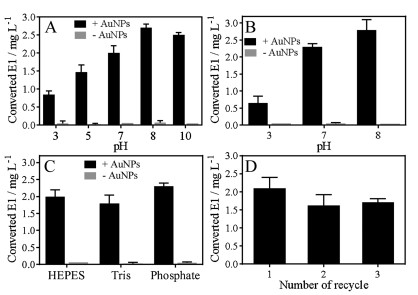
To further optimize and understand the reaction, we then mixed the AuNPs with E2 in different pH conditions and the results are summarized in the Fig. 3A. The yield of E2 oxidation product was higher at high pH. The maximum production of E1 was obtained at pH 8 reaching ~2.7 mg/L. As controls, basically no E1 was measured for all of the samples without AuNPs. We also tested the reaction in phosphate buffers of different pH and a higher activity was still observed at higher pH (Fig. 3B). If tested in different buffers but all at pH 7, the activities were similar (Fig. 3C). Therefore, the oxidation was mainly affected by pH and the type of buffer was not important.

|
Download:
|
| Fig. 3. Concentrations of obtained E1 from E2 oxidations catalyzed by the AuNPs in (A) different buffers with different pH, (B) phosphate buffers with different pH, (C) different buffers with same pH at 7. (D) The recycle tests of E2 oxidations catalyzed by AuNPs. E2 (0.1 mmol/L) and 13 nm AuNPs (5 nmol/L) were used for the reactions at room temperature. | |
{kind=link}
We reason that the reaction of E2 oxidation released protons so that producing E1 was favored at alkaline conditions. This phenomenon was also observed by using manganese oxide to catalyze E2 oxidation in soil [48]. In human metabolic pathway, dehydrogenase catalyzing E2 oxidation to E1 also produces protons that are taken by the enzyme cofactor NADP [49, 50]. An interesting comparison can be made is glucose oxidation catalyzed by AuNPs, where the AuNPs are mimics of glucose oxidase [20]. In this reaction, the oxidation rate is also higher at higher pH. Protons were produced during the reaction and the decrease of pH inhibited the activity [51]. Electrostatic force may be favorable for the catalysis, since E2 has a pKa of ~10.7 and it is positive charged in pH range [52], while the AuNPs are negative charged [53, 54]. Previous mechanism studies suggested that more active species were on gold surfaces compared with other metal nanomaterials. The reaction might follow the Eley–Rideal mechanism that the adsorbed E2 on the surface reacts with oxygen in the solution [20, 55].
The concentration of the AuNPs was 5 nmol/L. With a yield of 2.7 mg/L of E1 (10 μmol/L), each AuNP converted 2000 E2 to E1 during this reaction time of 20 min. Therefore, this is a quite efficient multi-turnover nanozyme. The AuNPs can be reused for multiple catalysis cycles by simple centrifugation and re-dispersion (Fig. 3D).
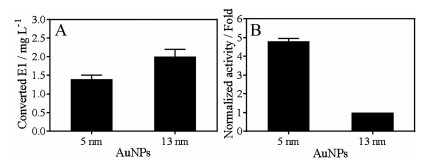
In general, smaller nanoparticles are more active due to larger specific surface area. In addition, smaller particles are more prone to form defect sites and these sites are often more catalytically active. We then used smaller AuNPs of 5 nm (Fig. S2 in Supporting information for TEM) to catalyze E2 oxidation and compared it with the 13 nm AuNPs. With same molar concentration (5 nmol/L), 5 nm AuNPs converted ~1.4 mg/mL of E1, which was slightly less than that of 13 nm AuNPs of ~2 mg/mL (Fig. 4A). Since the surface area of 5 nm AuNP is 6.7- fold smaller than that of a 13 nm AuNP, when comparing them based on same surface area, the oxidation yield of 5 nm AuNPs was ~4.8-fold higher than that of 13 nm AuNPs (Fig. 4B). Therefore, 5 nm AuNP is a more effective catalyst for E2 degradation likely due to it has a higher active site density.

|
Download:
|
| Fig. 4. (A) Concentration of converted E1 from E2 oxidation catalyzed by 5 nmol/L of 13 nm or 5 nm AuNPs. (B) The normalized activity based on the same surface area. For this experiment, 0.1 mmol/L E2 and 5 nmol/L AuNPs were used in HEPES buffer (20 mmol/L, pH 7) at room temperature. | |
{kind=link}
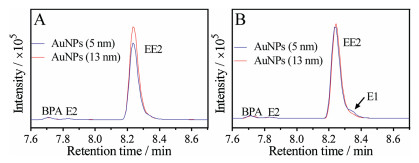
After confirming E2 oxidation catalyzed by the AuNPs, we then wanted to try EE2. We chose EE2 since it is a synthetic derivative of E2 and wildly used as estrogen medication. EE2 was mixed with the AuNPs for reaction and the products were analyzed by LC-MS in the same method (Fig. 5A). However, no products were observed indicating that EE2 did not degrade. Compared to E2, the carbon has an additional 17α-ethynyl group (Fig. 1A), making it a tertiary alcohol and more difficult to oxidize [46]. Next, we added H2O2 to the reaction. From the spectra (Fig. 5B), we found only a trace amount of E1 was produced with by 5 nm AuNPs while not for the 13 nm AuNPs. No E1 was found when only added H2O2 without AuNPs (Fig. S3 in Supporting information). In this case, the AuNPs were mimics of peroxidase activity in the reaction. Besides EE2, we also tried E3 and BPA as substrates for oxidation. However, no oxidation products were detected (Fig. S4 in Supporting information).

|
Download:
|
| Fig. 5. LC-MS spectra of EE2 oxidation catalyzed by 5 nm and 13 nm AuNPs (A) without, and (B) with 10 mmol/L H2O2 added in HEPES buffer (20 mmol/L, pH 7) at room temperature for 1 h. | |
{kind=link}
In conclusion, with more and more nanomaterials being produced for various applications, their environmental impact will have to be carefully studied. In this work, we screened six commonly used inorganic nanomaterials for reaction with the E2 estrogens. AuNPs were found capable of catalyzing E2 oxidation by simply mixing in aqueous solutions, while the tested metal oxide nanoparticles had no activity. The reaction has higher oxidation rate in alkaline conditions since the produced proton during oxidation. The AuNPs are mimics of dehydrogenases in human metabolic pathway for E2 degradation. The smaller size of AuNPs (e.g. 5 nm) has more effective catalytic activity due to its larger surface area for substrates accessibility. This work has expanded the scope of the nanozyme field by testing a new set of environmental substrates. The reaction may also be extended to some signal transduction mechanisms for biosensor development [56, 57]. We expect more developments in this front to show practical environmental and biological applications of nanozymes in the near future.
AcknowledgmentFunding for this work was from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.062.
| [1] |
C.J. Gruber, W. Tschugguel, C. Schneeberger, et al., N. Engl. J. Med. 346 (2002) 340-352. DOI:10.1056/NEJMra000471 |
| [2] |
H.G. Burger, Fertil. Steril. 77 (2002) 3-5. |
| [3] |
V.N. Luine, Horm. Behav. 66 (2014) 602-618. DOI:10.1016/j.yhbeh.2014.08.011 |
| [4] |
M. Adeel, X. Song, Y. Wang, et al., Environ. Int. 99 (2017) 107-119. DOI:10.1016/j.envint.2016.12.010 |
| [5] |
T.F.T. Omar, A. Ahmad, A.Z. Aris, et al., TrAC-Trends Anal. Chem. 85 (2016) 241-259. DOI:10.1016/j.trac.2016.08.004 |
| [6] |
R.N. Tanna, G.R. Tetreault, C.J. Bennett, et al., Environ. Toxicol. Chem. 32 (2013) 1981-1991. DOI:10.1002/etc.2262 |
| [7] |
M. Giulivo, M.L. de Alda, E. Capri, et al., Environ. Res. 151 (2016) 251-264. DOI:10.1016/j.envres.2016.07.011 |
| [8] |
C. Zhang, Y. Li, C. Wang, et al., Crit. Rev. Environ. Sci. Technol. 46 (2016) 1-59. DOI:10.1080/10643389.2015.1061881 |
| [9] |
F. Itzel, K.S. Jewell, J. Leonhardt, et al., Sci. Total Environ. 624 (2018) 1443-1454. DOI:10.1016/j.scitotenv.2017.12.181 |
| [10] |
A. Roudbari, M. Rezakazemi, AMB Express 8 (2018) 91. DOI:10.1186/s13568-018-0621-4 |
| [11] |
Z. Zhang, Y. Feng, Y. Liu, et al., J. Hazard. Mater. 181 (2010) 1127-1133. DOI:10.1016/j.jhazmat.2010.05.132 |
| [12] |
K. Sornalingam, A. McDonagh, J.L. Zhou, Sci. Total Environ. 550 (2016) 209-224. DOI:10.1016/j.scitotenv.2016.01.086 |
| [13] |
M.V. de Liz, R.M. de Lima, B. do Amaral, et al., J. Braz. Chem. Soc. 29 (2018) 380-389. |
| [14] |
Y. Huang, J. Ren, X. Qu, Chem. Rev. 119 (2019) 4357-4412. DOI:10.1021/acs.chemrev.8b00672 |
| [15] |
Y. Zhou, B. Liu, R. Yang, et al., Bioconj. Chem. 28 (2017) 2903-2909. DOI:10.1021/acs.bioconjchem.7b00673 |
| [16] |
J. Wu, X. Wang, Q. Wang, et al., Chem. Soc. Rev. 48 (2019) 1004-1076. DOI:10.1039/C8CS00457A |
| [17] |
S. Lin, H. Wei, Sci. China-Life Sci. 62 (2019) 710-712. DOI:10.1007/s11427-019-9518-0 |
| [18] |
G. Zhang, R. Wang, G. Li, Chin. Chem. Lett. 29 (2018) 687-693. DOI:10.1016/j.cclet.2018.01.043 |
| [19] |
W. Luo, C. Zhu, S. Su, et al., ACS Nano 4 (2010) 7451-7458. DOI:10.1021/nn102592h |
| [20] |
M. Comotti, C. Della Pina, R. Matarrese, et al., Angew. Chem. Int. Ed. 43 (2004) 5812-5815. DOI:10.1002/anie.200460446 |
| [21] |
L. Gao, J. Zhuang, L. Nie, et al., Nat. Nanotechnol. 2 (2007) 577-583. DOI:10.1038/nnano.2007.260 |
| [22] |
A. Asati, S. Santra, C. Kaittanis, et al., Angew. Chem. Int. Ed. 48 (2009) 2308-2312. DOI:10.1002/anie.200805279 |
| [23] |
M. Liu, Z. Li, Y. Li, J. Chen, Q. Yuan, Chin. Chem. Lett. 30 (2019) 1009-1012. DOI:10.1016/j.cclet.2018.12.021 |
| [24] |
B. Liu, J. Liu, Nano Res. 10 (2017) 1125-1148. DOI:10.1007/s12274-017-1426-5 |
| [25] |
Y. Hu, H. Cheng, X. Zhao, et al., ACS Nano 11 (2017) 5558-5566. DOI:10.1021/acsnano.7b00905 |
| [26] |
T. Yu, B. Liu, J. Liu, J. Anal. Test. 1 (2017) 2. DOI:10.1007/s41664-017-0001-0 |
| [27] |
L.Z. Gao, K.L. Fan, X.Y. Yan, Theranostics 7 (2017) 3207-3227. DOI:10.7150/thno.19738 |
| [28] |
X. Wang, Y. Hu, H. Wei, Inorg. Chem. Front. 3 (2016) 41-60. DOI:10.1039/C5QI00240K |
| [29] |
Z. Liu, X. Qu, Sci. China-Life Sci. 62 (2019) 150-152. DOI:10.1007/s11427-018-9417-1 |
| [30] |
L. Yu, Y. Chen, H. Chen, Chin. Chem. Lett. 28 (2017) 1841-1850. DOI:10.1016/j.cclet.2017.05.023 |
| [31] |
Z. Zhang, X. Zhang, B. Liu, et al., J. Am. Chem. Soc. 139 (2017) 5412-5419. DOI:10.1021/jacs.7b00601 |
| [32] |
B. Liu, Z. Huang, J. Liu, Nanoscale 8 (2016) 13562-13567. DOI:10.1039/C6NR02730J |
| [33] |
Y. Biniuri, B. Albada, M. Wolff, et al., ACS Catal. 8 (2018) 1802-1809. DOI:10.1021/acscatal.7b03454 |
| [34] |
M. Vazquez-Gonzalez, R.M. Torrente-Rodriguez, A. Kozell, et al., Nano Lett. 17 (2017) 4958-4963. DOI:10.1021/acs.nanolett.7b02102 |
| [35] |
M.S. Hizir, M. Top, M. Balcioglu, et al., Anal. Chem. 88 (2016) 600-605. DOI:10.1021/acs.analchem.5b03926 |
| [36] |
D. Li, B. Liu, P.J.J. Huang, et al., Chem. Commun. 54 (2018) 12519-12522. DOI:10.1039/C8CC07062H |
| [37] |
X.L. Ren, M.Q. Wang, X. He, et al., Chin. Chem. Lett. 29 (2018) 1865-1868. DOI:10.1016/j.cclet.2018.12.007 |
| [38] |
H. Liang, F. Lin, Z. Zhang, et al., ACS Appl. Mater. Interfaces 9 (2017) 1352-1360. DOI:10.1021/acsami.6b15124 |
| [39] |
C.I. Wang, W.T. Chen, H.T. Chang, Anal. Chem. 84 (2012) 9706-9712. DOI:10.1021/ac300867s |
| [40] |
J.H. Xu, C.G. Hu, S.S. Hu, Chin. Chem. Lett. 20 (2009) 1248-1250. DOI:10.1016/j.cclet.2009.04.019 |
| [41] |
B. Liu, J. Liu, ACS Appl. Mater. Interfaces 7 (2015) 24833-24838. DOI:10.1021/acsami.5b08004 |
| [42] |
M.J. Arlos, W.J. Parker, J.R. Bicudo, et al., Sci. Total Environ. 610 (2018) 1103-1112. |
| [43] |
M.J. Arlos, R. Liang, M.M. Hatat-Fraile, et al., J.Hazard.Mater. 318 (2016) 541-550. DOI:10.1016/j.jhazmat.2016.07.048 |
| [44] |
C. Della Pina, E. Falletta, L. Prati, et al., Chem. Soc. Rev. 37 (2008) 2077-2095. DOI:10.1039/b707319b |
| [45] |
T. Mallat, A. Baiker, Annu. Rev. Chem. Biomol. Eng. 3 (2012) 11-28. DOI:10.1146/annurev-chembioeng-062011-081046 |
| [46] |
H. Kuhl, Climacteric 8 (2005) 3-63. |
| [47] |
M.M. Miettinen, M.V. Mustonen, M.H. Poutanen, et al., Biochem. J. 314 (1996) 839-845. DOI:10.1042/bj3140839 |
| [48] |
G.D. Sheng, C. Xu, L. Xu, et al., Environ. Pollut. 157 (2009) 2710-2715. DOI:10.1016/j.envpol.2009.04.030 |
| [49] |
L.L. Engel, E.V. Groman, Human placental 17β-estradiol dehydrogenase: Characterization and structural studies, Proceedings of the 1973 Laurentian Hormone Conference, Elsevier, 1974, pp. 139-169.
|
| [50] |
R. Breton, D. Housset, C. Mazza, et al., Structure 4 (1996) 905-915. DOI:10.1016/S0969-2126(96)00098-6 |
| [51] |
N.J. Lang, B. Liu, J. Liu, J. Colloid Interface Sci. 428 (2014) 78-83. DOI:10.1016/j.jcis.2014.04.025 |
| [52] |
K. Lewis, R. Archer, Steroids 34 (1979) 485-499. DOI:10.1016/S0039-128X(79)80011-2 |
| [53] |
A. Kumar, S. Mandal, P. Selvakannan, et al., Langmuir 19 (2003) 6277-6282. DOI:10.1021/la034209c |
| [54] |
J. Liu, Phys. Chem. Chem. Phys. 14 (2012) 10485-10496. DOI:10.1039/c2cp41186e |
| [55] |
Y. Lin, J. Ren, X. Qu, Adv. Mater. 26 (2014) 4200-4217. DOI:10.1002/adma.201400238 |
| [56] |
Z. Qing, L. Zhu, S. Yang, et al., Biosens. Bioelectron. 78 (2016) 471-476. DOI:10.1016/j.bios.2015.11.057 |
| [57] |
D. Wang, B. Huang, J. Liu, et al., Biosens. Bioelectron. 102 (2018) 389-395. DOI:10.1016/j.bios.2017.11.051 |