 2019, Vol. 30
2019, Vol. 30 


b Hubei Cancer Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430079, China
In the central dogma of molecular biology, genetic information flows from DNA to RNA and then to proteins. In addition to the canonical nucleobases, plenty of chemical modifications have been identified presence in genomic DNA and RNA [1, 2]. These modifications do not alter the sequence context of nucleic acids, but change their biochemical and physical properties, and eventually control or regulate the spatiotemporal expression of genes [3]. Up to date, over 150 different types of chemical modifications have been found existence in nucleic acids [4, 5]. The modifications in nucleic acids play sophisticated roles in annotating genetic information [6, 7].
So far, seven modifications have been discovered in genomic DNA ofeukaryotes[7-9].5-Methylcytosine(m5C)is the most abundant and important epigenetic mark in DNA [10].m5C modification in DNA is dynamic and reversible and involves in numerous physiological processes[10].In2009, 5-hydroxymethylcytosine(hm5C), anoxidized derivative of m5C, was discovered in mammalian genome and TET (ten-eleven translocation) proteins were identified to be able to convert m5C to hm5C [11, 12]. Subsequent studies revealed that TET proteins can further oxidize hm5C to 5-formylcytosine (f5C) and 5-carboxycytosine (ca5C) [13, 14]. With these discovered modifications, these studies uncovered the mechanism of active DNA demethylation in mammals. Moreover, it was recently reported that DNA adenine methylation (N6-methyladenine, m6A) and the hydroxylation derivative of m6A were also present in genomic DNA of eukaryotes [9, 15-18]. These adenine modifications in DNA show crucial roles in regulating expression of genes [8].
In addition to DNA modifications, RNA molecules also carry various levels of modifications [19]. More than 150 different kinds of modifications exist in cellular RNA [2]. These modifications exert essential and critical functions in a great number of cellular processes in eukaryotic organisms, including affecting the stability, maturation, and secondary structures of RNA [19, 20]. Besides maintaining the higher structure and catalytic activity, RNA modifications also function as new information carrier in regulating cell physiology [19, 20].
Deciphering the biological roles of modifications in nucleic acids relies on the identification and location analysis of these modifications[21]. Methodsfor analyzing modifications in nucleic acids have tremendously advanced in the last decade [21-29]. Detection of nucleic acid modifications that relies on enzymatic digestion followed by liquid chromatography [30], thin layerchromatography (TLC) [31], and capillary electrophoresis [32], generally only provide the overall contents of modifications. Location information of these modifications in nucleic acids is essential and critical to reveal the functions of modifications. To map modifications in nucleic acids, some masss pectrometry(MS)-based analytical strategies have been developed [33]. Recent progress in next-generation sequencing (NGS) in conjugationwith immunoprecipitation, chemical reaction, or enzyme-mediated mutation allow genome-wide or transcriptome-wide mapping of modifications, which greatly revolutionize the field of nucleic acid modifications [34].
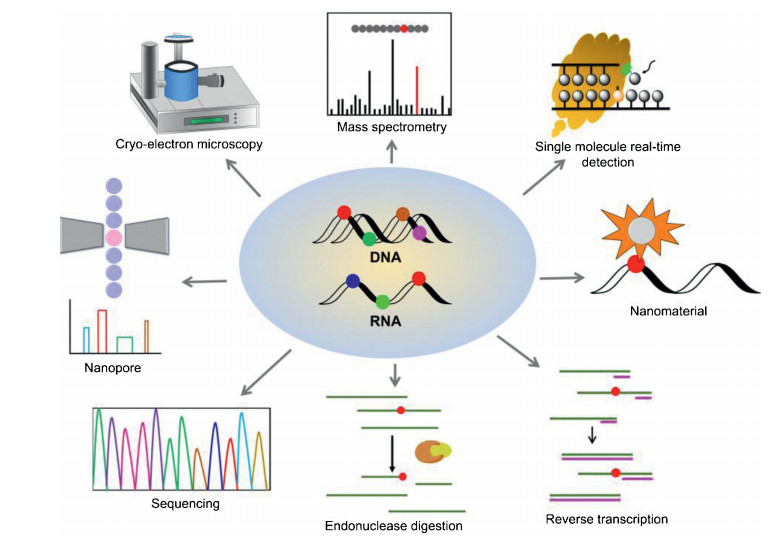
Herein, we review the established methods and the breakthrough of the techniques for location analysis of nucleic acid modifications. Fig. 1 is the summary of the analytical methods for location analysis of modifications in nucleic acids. We discuss the principles, applications, advantages and drawbacks of these methods. We believe that the advancement of techniques and methods will promote uncovering the functional roles of nucleic acid modifications.

|
Download:
|
| Fig. 1. Schematic illustration of the methods for the location analysis of modifications in nucleic acids. | |
2. Location analysis of modifications in nucleic acids 2.1. Mass spectrometry
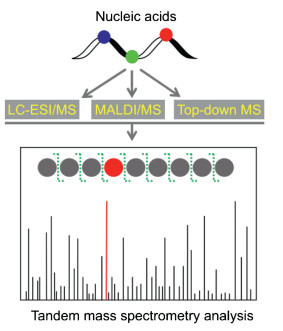
Mass spectrometry has superior capability in identification of compounds [35-39]. The rapid advances in MS-based analytical methods greatly promote the study of nucleic acid modifications and it becomes increasing important platform in the research field of nucleic acid modifications [40-46]. Tandem MS (MS/MS) is commonly performed to analyze the sequence of nucleic acids and modifications in nucleic acids [47]. Particularly, liquid chromatography-electrospray ionization/mass spectrometry (LC-ESI/MS), matrix-assisted laser desorption/ionization mass spectrometry (MALDI/MS), and top-down mass spectrometry (top-down MS) are the most widely used platforms for MS-base location analysis of modifications in nucleic acids (Fig. 2).

|
Download:
|
| Fig. 2. Schematic illustration of MS-based location analysis of modifications in nucleic acids. Nucleic acids generally are enzymatically digested to small fragments followed byLC-ESI/MSor MALDI/MS analysis. Top-downMS isable toanalyzeintact RNA without enzymatic digestion. Tandem MS analysis can provide the nuclei acid sequence information and location of modifications. | |
2.1.1. LC-ESI/MS
As nucleic acids typically are long polymers and have extremely large molecular weights, they generally are first enzymatically digested to small fragments followed by LC-ESI/MS analysis [48-54]. RNase T1 and RNase A are widely used in the digestion of RNA to form small oligonucleotide fragments, which were then subjected to LC-ESI/MS [55, 56]. Tandem MS (MS/MS) analysis can generate sequence information, including the sites of modifications on oligonucleotides [57, 58]. With this analytical strategy, the sites of various modifications in RNA were identified, including 3-methylpseudouridine (m3U) in 23S rRNA of E. coli [59], 5-methyluridine (m5U) and pseudouridine (ψ) in tmRNA of E. coli [60], 2-thiouridine (s2U) in mitochondria tRNA of L. tarentolae [61], 2-methylthio cyclic N6-threonylcarbamoyladenosine (ms2ct6A) at position 37 of tRNA from Bacillus subtilis, plants and Trypanosoma brucei [62]. LC-ESI/MS is only able to analyze short fragments of DNA and RNA, which limits its application on the genome-wide or transcriptome-wide mapping of modifications.
2.1.2. MALDI/MSIn addition to LC-ESI/MS, MALDI/MS is also frequently used for the analysis of nucleic acid fragments due to its capability on detection of relatively large molecular weight compounds. Prior to MALDI/MS analysis, RNA was also need to be fragmented by specific RNase, which are then analyzed by MALDI-MS/MS to locate the modifications [63]. The MALDI/MS was successfully employed in screening multiple modifications in rRNA [64].
ψ is a mass-silent modification and the normal MALDI/MS analysis cannot locate ψ in RNA. To address this issue, chemical labeling in combination with MALDI/MS analysis was developed for locating ψ modification in RNA [65]. In this method, 1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide (CMC) was used to selectively label ψ and the formed derivative of ψ carrying a 252 Da mass tag that can be easily identified by subsequent MALDI-MS/MS analysis [65]. In addition, the specific cyanoethylation of N1 in ψ by acrylonitrile was also utilized to locate ψ in RNA [66]. Cyanoethylated fragments can be readily identified by comparing the MS spectra of untreated and acrylonitrile-treated samples since the addition of one acrylonitrile leads to a mass increase of 53 Da. With this analytical strategy, ψ in E. coli tRNATyrII was identified. Cyanoethylation is a useful complement to the CMC labeling on ψ location analysis.
2.1.3. Top-down MSAn alternative strategy for the location analysis of modifications in nucleic acids is through the top-down MS analysis of the intact RNA without enzymatic digestion. For example, top-down MS was successfully used for the identification of the presence of 2'-O-methylation on the terminal of microRNA in plant [67]. With the top-down MS analysis, 89% sequence coverage in a single collisionally activated dissociation spectrum of E. coli tRNAVal was obtained and many modifications in tRNA also can be located [68]. Very recently, top-down MS was used to characterize RNA methylations (m6A, m5C, m3U, and m5U) in the mixture of either isomers of RNA or non-isomeric RNA forms [69]. The top-down MS-based analysis relies on advanced mass spectrometers. Right now only pure RNA samples can be analyzed by top-down MS. Thus, this method still needs great improvement for locating modifications in nucleic acids from biological samples.
2.2. SequencingSequencing can provide comprehensive information of nucleotides in DNA and RNA. In the last several years, many powerful strategies based on sequencing in conjugation with a variety of approaches have been developed and established for genomewide and transcriptome-wide mapping of a variety of modifications in nucleic acids.
2.2.1. Immunoprecipitation-mediated sequencingNucleic acid modifications generally are present in lowabundance. Therefore, the enrichment of the fragments that carry modifications will benefit the subsequent sequencing for mapping modifications. Immunoprecipitation-mediated enrichment of nucleic acid fragments using antibodies or affinity binding proteins has been demonstrated to be very useful for mapping modifications in nucleic acids [70]. The drawback of immunoprecipitation-mediated sequencing is that this strategy generally cannot offer location information of modifications at single-base resolution.
Many immunoprecipitation-mediated sequencing, such as methylated DNA binding domain sequencing (MBD-seq) [71], methylation DNA immunoprecipitation sequencing (MeDIP-seq) [72], MIRA-seq [73], MethylCap-seq [74], and m6A DIP-seq [16, 75, 76] have been established for the comprehensive mapping of m5C and m6A in genomic DNA. In addition, hm5C DNA immunoprecipitation-sequencing using antibodies against hm5C [77, 78], or antibody against cytosine 5-methylenesulfonate (CMS) [79] were also developed to map hm5C in genome. These mapping studies demonstrated that hm5C was mainly enriched in enhancers, promoters, and gene bodies, revealing a critical role of hm5C in regulation of gene expressions. Similarly, immunoprecipitation-mediated enrichment of f5C followed by NGS realized the genome-wide mapping of f5C [80].
In addition to DNA modifications, immunoprecipitation of RNA fragments carrying modifications followed by NGS (RIP-seq) offers a promising platform for transcriptome-wide mapping of RNA modifications. In recent years, m6A-specific RNA immunoprecipitation coupled with NGS (MeRIP-seq) was developed for transcriptome-wide mapping of m6A, which provided an effective approach for locating m6A in mRNA and revealed the regulatory roles of m6A [81-83]. However, MeRIP-seq only can map m6A in RNA at a resolution of approximately 100-200 nt. Recently, crosslinking and immunoprecipitation (miCLIP) in conjugation NGS analysis was developed for mapping m6A at single-base-resolution [84]. Moreover, the m1A-ID-seq [85, 86] and ac4C-RIP-seq [87] for transcriptome-wide mapping of m1A and N4-acetylcytidine (ac4C), respectively, based on immunoprecipitation-mediated sequencing were also established. Collectively, these approaches allow comprehensive location analysis of modifications and provide valuable platforms for functional studies of modifications in nucleic acids.
2.2.2. Chemical reaction-mediated sequencingThe bisulphite-converted strategy is the gold standard for mapping m5C in DNA. Bisulphite sequencing can provide single-base resolution of m5C in genomic DNA [88]. After the bisulphite treatments, the genome is largely composed of only three nucleotides (A, G, and T). Thus, methods relying on bisulphite sequencing need extensive bioinformatics analysis.
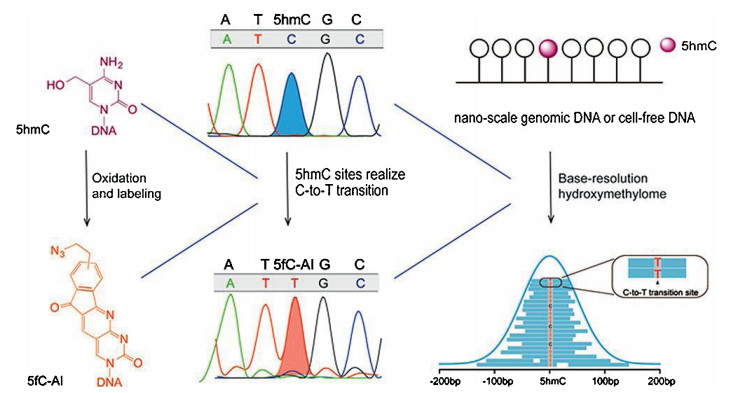
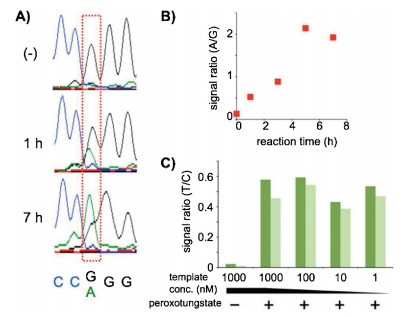
Inadditionto m5C inDNA, oxidative bisulphite sequencing(oxBS-seq)strategy was developed for mappinghm5C inDNA atsingle-base resolution [89]. The oxBS-seq approach is based on the specific oxidation of hm5C to form f5C by potassium perruthenate (KRuO4), which enables conversion of the formed f5C to uracil by bisulphite treatment. The hm5C would therefore read as thymine, whereas m5C would still read as cytosine in DNA treated with KRuO4 by subsequent bisulphite sequencing. By comparing the sequencing results obtained by oxBS-seq and traditional bisulphite sequencing, the hm5C sites can be obtained at single-baseresolution.Morerecently, a method named hmC-CATCH that was based on selective hm5C oxidation by potassium ruthenate (K2RuO4), chemical labeling and subsequentC-to-Ttransition during PCR, was developed for mapping hm5C in genomes (Fig. 3) [90]. This method required only nanoscale genomic DNA, which could greatly benefit the hm5C mapping analysis with limited biological and clinical samples. In addition, peroxotungstate-mediated oxidation and sequencing analysis was developed for mapping hm5C at single-base resolution [91]. In this strategy, the oxidized productof hm5C, trihydroxylated thymine, can pair with dATP. Bycomparing the sequencing results before and after the peroxotungstate oxidation, hm5C sites in DNA could be specifically located (Fig. 4).

|
Download:
|
| Fig. 3. Schematic illustration for mapping hm5C in RNA at single-base resolution. This method consists of hm5C selective oxidation by potassium ruthenate (K2RuO4), chemical labeling and subsequent C-to-T transition during sequencing. 1, 3-Indandione (AI) was used to label the oxidized product of hm5C (i.e., f5C) to form f5C-AI that will be read as T during sequencing. This method required only nanoscale genomic DNA. Copied with permission [90]. Copyright 2018, American Chemical Society. | |

|
Download:
|
| Fig. 4. TUC-seq analysis of hm5C in DNA. (A) Sequencing data before and after 1 or 7 h oxidation of hm5C-DNA. (B) Oxidation-time-dependent change of A/G signal ratio. (C) Template concentration dependence of T/C signal ratio of PCR-amplified samples derived from hm5C-DNA. The ratio values derived from the 5'-and 3'-side hm5C positions are shown in dark and light green, respectively. Copied with permission [91]. Copyright 2016, American Chemical Society. | |
Moreover, a strategy of TET-assisted bisulphite sequencing (TAB-seq) was also developed for single-base resolution mapping of hm5C [92]. In TAB-seq appraoch, hm5C is glycosylated by β-glucosyltransferase (β-GT), which prevented from TET-mediated oxidation. Then TET enzyme treatment causes the oxidation of m5C and f5C to ca5C, which were read as thymine in subsequent bisulphite sequencing. The remaining cytosine signals are then from the protected hm5C. The advantage of TAB-seq is that there is no need for the comparison with traditional bisulphite sequencing and therefore hm5C can be identified directly.
Recently, a reduced bisulphite sequencing (redBS-seq) method was established to map f5C in DNA at single-base resolution [93].This redBS-seq is based on a selective reduction of f5C to hm5C followed by bisulphite sequencing. By combining redBS-seq and oxBS-seq, the genome-wide mapping of m5C, hm5C and f5C in mouse embryonic stem cells was achieved. In addition, Yi's group developed a bisulfite-free method for genome-wide analysis of f5C based on 1, 3-indandione labeling of f5C by and subsequent C-to-T transition in sequencing [94]. This strategy shows no noticeable DNA degradation and provides single-base resolution mapping of 5f C. As for ca5C, a chemical modification-assisted bisulphite sequencing (CAB-seq) was developed for locating ca5C in DNA at single-base resolution [95]. The carboxyl group of ca5C was labeled by the compound containing primary amine group, which endows the resistance of labeled ca5C to deamination by bisulphite treatment. Therefore, ca5C can be distinguished from other nucleobases during bisulphite sequencing.
Similarly, bisulfite-mediated sequencing was also reported for the location analysis of m5C in both rRNA and tRNA of Drosophila [96], m5C in mRNA of human cells [97] and in mRNA of S. solfataricus [98]. Since bisulphite treatment may result in substantial degradation of nucleic acids and RNA sample is more prone to degradation than DNA, the bisulphite treatment reaction need to be carefully optimized while analysis of RNA samples.
Alkaline hydrolysis followed by NGS analysis (RiboMeth-seq) was developed to map 2′-O-methylation sites in yeast rRNA [99]. Under alkaline condition, the ribose sugar with 2'-OH can be cleaved; however, the 2′-O-methylated ribose is resistant to alkaline cleavage. The differential properties of 2′-O-methylated and 2'-OH ribose were then utilized to map 2′-O-methylation in rRNA [100]. More recently, periodate oxidation reaction was also utilized to establish a method of Nm-seq for transcriptome-wide mapping of 2′-O-methylation at single-base-resolution by virtue of the differential reactivity of 2′-O-methylated and 2'0-OH nucleotides to periodate oxidation [101]. In addition, a method of thiouridine-to-cytidine-sequencing (TUC-seq) was developed for mapping 4-thiouridine (s4U) in RNA [102]. The reaction of s4U with osmium tetroxide (OsO4) and ammonia can result in conversion of s4U into cytosine, which was then utilized for a comparative sequencing analysis. For untreated RNA, s4U is read as T while for OsO4-treated RNA, s4U is read as C. This TUC-seq enabled the location analysis of s4U in RNA at single-base resolution.
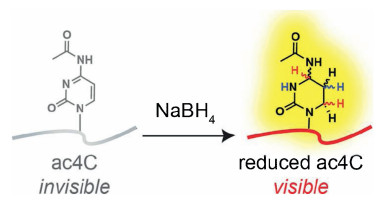
Adenosine-to-inosine (A-to-I) editing is prevalent in RNA [103]. However, inosine cannot be directly detected by reverse transcription due to its base-pair with cytidine. Specifically chemical labeling with modifications may change their reverse transcription (RT) signature by RT arrest or misincorporation. In this respect, inosine chemical erasing (ICE) approach was established to map inosines in mRNA based on cyanoethylation reaction [104]. In ICE-seq, acrylonitrile was used to cyanoethylate inosine to form N1-cyanoethylinosine (ce1I), which can compromise the base-pairing with cytidine and induce the RT stop. This unique signature can be obtained by subsequent NGS analysis. With this ICE-seq, 5072 inosine sites in mRNA of human brain tissue were identified [104], which provides a powerful approach to decipher the function of A-to-I editing. Similarly, the borohydride-based reduction of ac4C in RNA can induce the RT misincorporation, which provided a unique chemical signature and was successfully applied for the location analysis of ac4C in RNA (Fig. 5) [105]. In addition, an approach of N3-CMC-enriched ψ sequencing (CeU-seq) was also developed to map ψ in mRNA of human cells [106]. In CeU-seq, N3-CMC was used to specifically react with ψ on RNA followed by subsequent enrichment and NGS analysis. The CMC-ψ complex can cause the RT stop one base downstream of ψ, which can provide transcriptomewide mapping of ψ at single-base-resolution. Similarly, iodoacetamide (IAA) was used to specifically react with s4U to form alkylated s4U that preferably pairs with G than A and therefore causes the T-to-C mutation [107]. This signature of IAA labeled s4U can be used for map s4U in RNA at single-base resolution.

|
Download:
|
| Fig. 5. Schematic illustration for mapping ac4C in RNA at single-base resolution. Chemical reduction of ac4C by NaBH4 to tetrahydro-ac4C. Tetrahydro-ac4C produces a unique mutational signaturethat can be applied to map ac4C in RNA at single-base resolution. Copied with permission [105]. Copyright 2018, American Chemical Society. | |
2.2.3. Enzyme-mediated sequencing
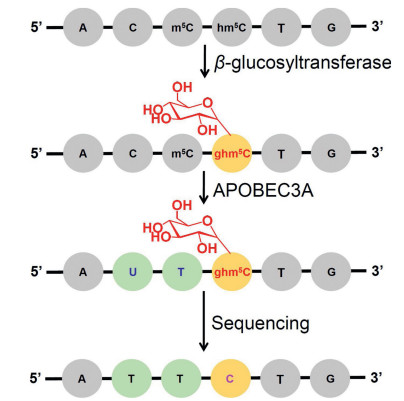
Our group and others recently reported an enzyme-mediated sequencing strategy for mapping hm5C in DNA [108, 109]. Previous report demonstrated that cytosine deaminases of APOBEC3A (apolipoproteinBmRNA-editing catalytic polypeptide-like 3 A)can deaminate cytosine to form uracil [110]. We further identified that APOBEC3A can effectively deaminate C, m5C, and hm5C, but show no observable deamination activity toward glycosylated hm5C (β-glucosyl-5-hydroxymethyl-2'-deoxycytidine, ghm5C). With this unique property of APOBEC3A, we established the APOBEC3 A-mediated deamination sequencing (AMD-seq) for location analysis of hm5C in DNA at single-base resolution (Fig. 6). In AMD-seq, APOBEC3A deaminate C and m5C in DNA to form U and T; while ghm5C is resistant to APOBEC3 Adeamination and will still read as C during sequencing (Fig. 7). Thus, the detected remaining C in DNA by sequencing could only come from original hm5C, which provides the single-base resolution analysis of hm5C. Since the AMD-seq analytical strategy does not need bisulphite treatment, the significant degradation of DNA can be avoided, which benefit the highlysensitive mappingof hm5C in DNAeven at single cell level.

|
Download:
|
| Fig. 6. The schematic illustration of the single-base resolution analysis of hm5C in DNA by AMD-seq. A glycosyl group is added to hm5C to form ghm5C that is resistant to APOBEC3A-mediated deamination. The ghm5C will read as C during sequencing, which can be used to map hm5C in DNA. Copied with permission [108]. Copyright 2018, American Chemical Society. | |

|
Download:
|
| Fig. 7. Sequencing analysis of APOBEC3 A-treated DNA. The sequencing results of (A) L-DNA-C, (B) L-DNA-m5C, (C) L-DNA-hm5C, and (D) L-DNA-ghm5C with or without APOBEC3 A treatment. Copied with permission [108]. Copyright 2018, American Chemical Society. | |
2.2.4. Reverse transcription-mediated sequencing
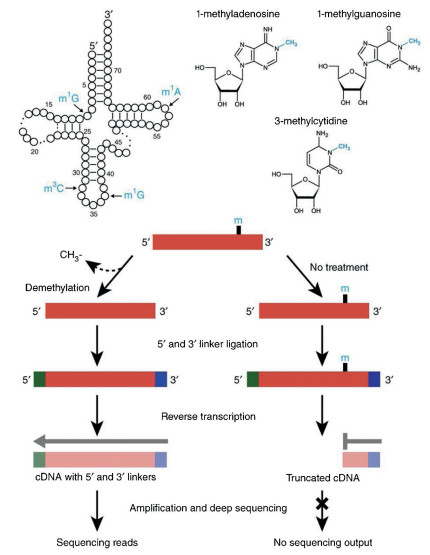
The RT-mediated arrest or misincorporation in cDNA sequences can be analyzed by subsequent NGS. For example, AlkBfacilitated RNA methylation sequencing (ARM-seq) utilized the property that m1A, m1G, and m3C can cause RT-arrest to map their sites in tRNA (Fig. 8) [111]. In ARM-seq, the full-length cDNA can be obtained using AlkB-treated RNA since AlkB removed the methyl group of m1A, m1G, and m3C. However, the native RNA without AlkB treatment only produced truncated cDNA due to RTarrest caused by these modifications. Thus, the comparison of NGS results in treated and untreated samples can provide the modification sites in RNA. Recently, an antibody-independent method to map m6A at single-nucleotide resolution by 4SedTTP incorporation was established [112]. 4SedTTP can form stable base pair with adenine but not with m6A. Therefore, the use of the 4SedTTP instead of TTP in reverse transcription led to the RT stop at m6A sites, which was then utilized to locate m6A in RNA. In addition, an engineered RT-active KlenTaq DNA polymerase variant was employed for mapping m6A in RNA [113]. The KlenTaq DNA polymerase variant exhibited increased misincorporation opposite m6A compared to adenine, which allowed the direct identification of m6A sites from the sequencing data. However, the RT-mediated sequencing for mapping modifications in RNA can be largely affected by the RNA higher structures. Thus, other methods are essential to confirm the results obtained by RT-mediated sequencing analysis.

|
Download:
|
| Fig. 8. Schematic illustration for mapping modifications in tRNA by ARM-seq. ARMseq utilized the property that m1A, m1G, and m3C can cause RT-arrest to map their sites in tRNA. The full-length cDNA can be obtained using AlkB-treated RNA since AlkB removed the methyl group of m1A, m1G, and m3C. However, the native RNA without AlkB treatment only produced truncated cDNA due to RT-arrest caused by these modifications. The comparison of sequencing results in treated and untreated samples can provide the modification sites in RNA. Copied with permission [111]. Copyright 2015, Springer Nature Publishing AG. | |
2.3. Endonuclease digestion-mediated location analysis
Some endonucleases can recognize and cleave specific modifications in RNA. The produced patterns can offer the location information of modifications by subsequent PCR, TLC, or gel electrophoresis analysis. Glyoxal can form stable complex with G, but not inosine, which therefore was used to map inosine sites in RNA [114]. Upon glyoxal treatment, the formed glyoxalG-borate complex is resistant to RNase T1 cleavage and only inosine sites were cleaved by RNase T1. The resulting fragments were then analyzed by gel electrophoresis, which provides a way to locate inosines in RNA. In addition, the RNase H cleavage in combination with gel electrophoresis analysis has been successfully employed in the location analysis of ψ in RNA [115], 2′-O-methylguanosine in 18S rRNA and 2′-O-methyladenosine in 28S rRNA of Xenopus [116].
Endonuclease digestion-mediated location analysis generally requires relatively large amount of purified RNA, which causes the analysis of modifications in individual mRNA or lncRNA challenging. Recently, a method of site-specific cleavage and radioactivelabeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET) was developed to map the sites of m6A in mRNA and lncRNA [117]. In the SCARLET method, RNase H digestion, ligation, and TLC analysis were sequentially carried out. With the SCARLET method, the m6A sites in two human lncRNA and three mRNA were successfully identified. SCARLET method requires no expensive instruments and can be easily applicable for mapping various modifications in RNA.
2.4. Reverse transcription-mediated location analysisA number of modifications in RNA can lead to RT arrest or misincorporation of dNTP, which can be used as characterized signature for locating modifications in RNA by subsequent PCR or gel electrophoresis analysis [118]. For example, some modifications in RNA were specifically mapped by RT-mediated arrest or misincorporation, such as m1G in E. coli 23S rRNA [119], N6, N6- dimethyladenosine in yeast 18S rRNA [120], 2′-O-methylation in mouse piwi-interacting RNA (piRNA) and in yeast and human rRNA [121].
Chemical reagents that can selectively react with certain modification can be used to label the modification and then enhance the signature of RT-mediated arrest or misincorporation. In this respect, CMC (N-cyclohexyl-N′-β-(4-methylmorpholinium) ethylcarbodiimide p-tosylate) was utilized to modify ψ, U and G [122]. After labeling, only CMC-ψ complex is stable, which therefore induced the RT arrest at ψ. Application of this method enabled the identification of four ψ sites in E. coli 23S RNA.
2.5. Single molecule real-time detectionThe innovative third-generation sequencing technologies of single molecule real-time detection (SMRT) can realize the direct location analysis of m5C and m6A in DNA [123-125]. In SMRT sequencing, DNA polymerases catalyze the incorporation of nucleotides into DNA. The discrimination of different nucleobase in DNA can be obtained by the unique kinetic signatures of each nucleotide. In addition to m5C and m6A, chemical labeling-mediated SMRT sequencing also achieved the mapping of hm5C in genomic DNA of mouse embryonic stem cells at single-base resolution [126].
In addition to DNA, modifications in RNA also can be mapped by SMRT sequencing [127]. The kinetics of reverse transcriptase that synthesizes cDNA can be monitored to obtain the sequence content and sites of modifications in RNA. The kinetic signature of reverse transcriptase on the m6A is obviously different with other nucleobases, which was then successfully used to locate m6A [127]. However, the location analysis of modifications in RNA by SMRT currently has only been achieved in synthetic RNA, and great effort is still needed to realize the location analysis of modifications in cellular RNA by SMRT.
2.6. NanoporeNanopore sequencing has emerged as a promising approach in location analysis of modifications in nucleic acids. The characterized ionic current while molecules pass through the nanoscale pore can be measured and utilized to differentiate modified nucleosides from normal nucleosides [128]. Nanopore sequencing is able to readily distinguish m5C from cytosine in DNA [129, 130]. m5C and hm5C also can be easily discriminated in DNA molecules by nanopore sequencing [131]. Moreover, chemical labeling of hm5C can improve the performance of nanopore sequencing for locating hm5C [132, 133]. Modifications in RNA, such as m6A, m5C, and inosine also can be distinguished by nanopore sequencing (Fig. 9) [134]. The nanopore sequencing shows to be promising and is expected to have a transformative impact on nucleic acid modifications study.

|
Download:
|
| Fig. 9. Schematic illustration for location analysis of modifications in RNA by nanopore sequencing. Copied with permission [134]. Copyright 2012, American Chemical Society. | |
2.7. Cryo-electron microscopy
The fast advancement of cryo-electron microscopy technology makes it become an important tool for deciphering the structures of biological specimens [135]. Recently, over 130 chemical modifications were visualized in rRNA of HeLa cells by cryoelectron microscopy analysis (Fig. 10) [136]. The high resolution of approximately 2.5Å allowed the distinct determination of the chemical structures of the rRNA. In addition, cryo-electron microscopy can provide detailed structures of the modifications in rRNA, which led to the discovery of many new types and sites of modifications in rRNA. It is believed that the powerful cryoelectron microscopy may play increasing important role and promote the in-depth investigation of nucleic acid modifications.

|
Download:
|
| Fig. 10. Modifications identified in the 60S rRNA by cryo-electron microscopy. Copied with permission [136]. Copyright 2017, Springer Nature Publishing AG. | |
2.8. Nanomaterials
Ligase chain reaction in combination with gold nanoparticle (AuNP)-based colorimetric assay was developed for the location analysis of m5C in DNA at one-base resolution [137]. In this strategy, DNA was treated with bisulfite, where the m5C in DNA remained unchanged and cytosine in DNA was converted to uracil. Then the bisulfite-treated DNA was used as the template for the ligation of the two pieces of complementary strands. The ligation position was designed at the site of m5C. Thus, the ligated products can be detected by subsequent ligase chain reaction and AuNPs aggregation induced red-to-blue color change, which allowed the visual detection of m5C in DNA with the naked eye. In addition to AuNPs, the quantum dot nanomaterials also was employed in conjugation with ligase chain reaction for the location analysis of m5C in DNA [138]. This kind of nanomaterial-based detection generally can determine m5C in DNA at single site with high detection limit.
3. Conclusions and perspectivesInnovations in technologies and analytical methods have greatly promoted the location study of nucleic acid modifications. In this review, we discussed the principles, advantages and drawbacks of the analytical strategies for mapping nucleic acid modifications and we summarized the important features of these methods in Table 1. Although MS exhibits particular capability on the discovery of new modifications, MS-based analysis may continue to play an important role in the location analysis of nucleic acid modifications. The fast advances of NGS-based methods are becoming the routine tools in study of nucleic acids. Further investigation of a variety of chemical reactions to be coupled with NGS is a promising direction for mapping modifications in nucleic acids. Many chemical reactions can be explored and new reagents can be synthesized to selectively label specific modifications in DNA or RNA.
|
|
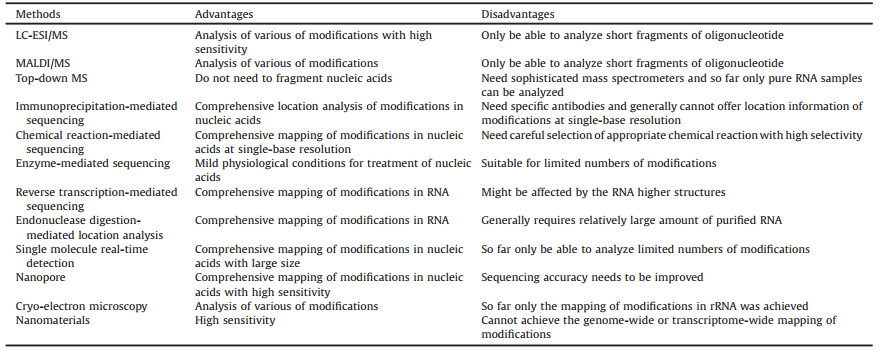
Table 1 Advantages and disadvantages of the methods for mapping modifications in nucleic acids. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Innovative third-generation sequencing technologies show extraordinary promising on location analysis of modifications. SMRT and nanopore sequencing provide the useful platforms for direct mapping modifications occurring in nucleic acids without additional chemical or enzymatic treatment. Nanopore sequencing has been well applied in location study of DNA modifications; while the application of third-generation sequencing technologies for locating modifications in RNA from real biological samples may still take some time.
Identification of modifications and mapping their sites in nucleic acids remain challenging. The advancement of technologies and new methods may lead to the discovery of novel modifications. By understanding the property of each analytical method, researchers can select the most suitable strategy for mapping these modifications in nucleic acids. And we expect that new advancement of techniques will promote the mapping study of modifications in nucleic acids and eventually improve our understanding of these modifications.
AcknowledgmentsThe authors thank the National Key R&D Program of China (No.2017YFC0906800) and the National Natural Science Foundation of China (Nos. 21672166, 21635006, 21721005, 21728802) for the financial support.
| [1] |
T. Carell, M.Q. Kurz, M. Muller, M. Rossa, F. Spada, Angew. Chem. Int. Ed. Engl. 57 (2018) 4296-4312. DOI:10.1002/anie.201708228 |
| [2] |
P. Boccaletto, M.A. Machnicka, E. Purta, et al., Nucleic Acids Res. 46 (2018) D303-D307.
|
| [3] |
K. Chen, B.S. Zhao, C. He, Cell Chem. Biol. 23 (2016) 74-85. DOI:10.1016/j.chembiol.2015.11.007 |
| [4] |
B. Chen, B.F. Yuan, Y.Q. Feng, Anal. Chem. 91 (2019) 743-756. DOI:10.1021/acs.analchem.8b04078 |
| [5] |
T. Liu, C.J. Ma, B.F. Yuan, Y.Q. Feng, Sci. China Chem. 61 (2018) 381-392. DOI:10.1007/s11426-017-9186-y |
| [6] |
J. Song, C. Yi, ACS Chem. Biol. 12 (2017) 316-325. DOI:10.1021/acschembio.6b00960 |
| [7] |
X. Wu, Y. Zhang, Nat. Rev. Genet. 18 (2017) 517-534. |
| [8] |
G.Z. Luo, C. He, Nat. Struct. Mol. Biol. 24 (2017) 503-506. DOI:10.1038/nsmb.3412 |
| [9] |
J. Xiong, T.T. Ye, C.J. Ma, et al., Nucleic Acids Res. 47 (2019) 1268-1277. DOI:10.1093/nar/gky1218 |
| [10] |
X. Lu, B.S. Zhao, C. He, Chem. Rev. 115 (2015) 2225-2239. DOI:10.1021/cr500470n |
| [11] |
S. Kriaucionis, N. Heintz, Science 324 (2009) 929-930. DOI:10.1126/science.1169786 |
| [12] |
M. Tahiliani, K.P. Koh, Y. Shen, et al., Science 324 (2009) 930-935. DOI:10.1126/science.1170116 |
| [13] |
S. Ito, L. Shen, Q. Dai, et al., Science 333 (2011) 1300-1303. DOI:10.1126/science.1210597 |
| [14] |
Y.F. He, B.Z. Li, Z. Li, et al., Science 333 (2011) 1303-1307. DOI:10.1126/science.1210944 |
| [15] |
E.L. Greer, M.A. Blanco, L. Gu, et al., Cell 161 (2015) 868-878. DOI:10.1016/j.cell.2015.04.005 |
| [16] |
Q. Xie, T.P. Wu, R.C. Gimple, et al., Cell 175 (2018) 1228-1243. DOI:10.1016/j.cell.2018.10.006 |
| [17] |
G. Zhang, H. Huang, D. Liu, et al., Cell 161 (2015) 893-906. DOI:10.1016/j.cell.2015.04.018 |
| [18] |
T.P. Wu, T. Wang, M.G. Seetin, et al., Nature 532 (2016) 329-333. DOI:10.1038/nature17640 |
| [19] |
I.A. Roundtree, M.E. Evans, T. Pan, C. He, Cell 169 (2017) 1187-1200. DOI:10.1016/j.cell.2017.05.045 |
| [20] |
S. Li, C.E. Mason, Annu. Rev. Genomics Hum. Genet. 15 (2014) 127-150. DOI:10.1146/annurev-genom-090413-025405 |
| [21] |
M. Helm, Y. Motorin, Nat. Rev. Genet. 18 (2017) 275-291. |
| [22] |
M.D. Lan, B.F. Yuan, Y.Q. Feng, Chin. Chem. Lett. 30 (2019) 1-6. DOI:10.1016/j.cclet.2018.04.021 |
| [23] |
B.F. Yuan, Y.Q. Feng, TrAC-Trend. Anal. Chem. 54 (2014) 24-35. DOI:10.1016/j.trac.2013.11.002 |
| [24] |
Y. Tang, J.M. Chu, W. Huang, et al., Anal. Chem. 85 (2013) 6129-6135. DOI:10.1021/ac4010869 |
| [25] |
Y. Tang, J. Xiong, H.P. Jiang, et al., Anal. Chem. 86 (2014) 7764-7772. DOI:10.1021/ac5016886 |
| [26] |
Y. Tang, S.J. Zheng, C.B. Qi, Y.Q. Feng, B.F. Yuan, Anal. Chem. 87 (2015) 3445-3452. DOI:10.1021/ac504786r |
| [27] |
Q.Y. Li, B.F. Yuan, Y.Q. Feng, Chem. Lett. 47 (2018) 1453-1459. |
| [28] |
W. Huang, M.D. Lan, C.B. Qi, et al., Chem. Sci. 7 (2016) 5495-5502. DOI:10.1039/C6SC01589A |
| [29] |
H. Zeng, C.B. Qi, T. Liu, et al., Anal. Chem. 89 (2017) 4153-4160. DOI:10.1021/acs.analchem.7b00052 |
| [30] |
M. Buck, M. Connick, B.N. Ames, Anal. Biochem. 129 (1983) 1-13. DOI:10.1016/0003-2697(83)90044-1 |
| [31] |
H. Grosjean, G. Keith, L. Droogmans, Methods Mol. Biol. 265 (2004) 357-391. |
| [32] |
M.G. Cornelius, H.H. Schmeiser, Electrophoresis 28 (2007) 3901-3907. DOI:10.1002/elps.200700127 |
| [33] |
C. Wetzel, P.A. Limbach, Analyst 141 (2016) 16-23. DOI:10.1039/C5AN01797A |
| [34] |
P.A. Limbach, M.J. Paulines, Wiley Interdiscip. Rev. RNA 8 (2017) e1367.
|
| [35] |
T. Huang, M.R. Armbruster, J.B. Coulton, J.L. Edwards, Anal. Chem. 91 (2018) 109-125. DOI:10.1021/acs.analchem.7b04669 |
| [36] |
N. Hamada, Y. Hashi, S. Yamaki, et al., Chin. Chem. Lett. 30 (2019) 99-102. DOI:10.1016/j.cclet.2018.10.029 |
| [37] |
Z. Wang, L. Chi, Chin. Chem. Lett. 29 (2018) 11-18. DOI:10.1016/j.cclet.2017.08.050 |
| [38] |
W. Huang, C.B. Qi, S.W. Lv, et al., Anal. Chem. 88 (2016) 1378-1384. DOI:10.1021/acs.analchem.5b03962 |
| [39] |
W. Huang, J. Xiong, Y. Yang, et al., RSC Adv. 5 (2015) 64046-64054. DOI:10.1039/C5RA05307B |
| [40] |
S. Liu, Y. Wang, Chem. Soc. Rev. 44 (2015) 7829-7854. DOI:10.1039/C5CS00316D |
| [41] |
J.M. Chu, T.T. Ye, C.J. Ma, et al., ACS Chem. Biol. 13 (2018) 3243-3250. DOI:10.1021/acschembio.7b00906 |
| [42] |
J. Xiong, X. Liu, Q.Y. Cheng, et al., ACS Chem. Biol. 12 (2017) 1636-1643. DOI:10.1021/acschembio.7b00170 |
| [43] |
J.M. Chu, C.B. Qi, Y.Q. Huang, et al., Anal. Chem. 87 (2015) 7364-7372. DOI:10.1021/acs.analchem.5b01614 |
| [44] |
H.P. Jiang, T. Liu, N. Guo, et al., Anal. Chim. Acta 981 (2017) 1-10. DOI:10.1016/j.aca.2017.06.009 |
| [45] |
H.Y. Zhang, J. Xiong, B.L. Qi, Y.Q. Feng, B.F. Yuan, Chem. Commun. (Camb.) 52 (2016) 737-740. DOI:10.1039/C5CC07354E |
| [46] |
H.P. Jiang, J. Xiong, F.L. Liu, et al., Chem. Sci. 9 (2018) 4160-4167. DOI:10.1039/C7SC05472F |
| [47] |
A. Nyakas, S.R. Stucki, S. Schurch, J. Am. Soc. Mass. Spectrom. 22 (2011) 875-887. DOI:10.1007/s13361-011-0106-z |
| [48] |
J.A. Kowalak, S.C. Pomerantz, P.F. Crain, J.A. McCloskey, Nucleic Acids Res. 21 (1993) 4577-4585. DOI:10.1093/nar/21.19.4577 |
| [49] |
C.B. Qi, H.P. Jiang, J. Xiong, B.F. Yuan, Y.Q. Feng, Chin. Chem. Lett. 30 (2019) 553-557. DOI:10.1016/j.cclet.2018.11.029 |
| [50] |
M.D. Lan, J. Xiong, X.J. You, et al., Chem.-Eur. J. 24 (2018) 9949-9956. DOI:10.1002/chem.201801640 |
| [51] |
M.L. Chen, F. Shen, W. Huang, et al., Clin. Chem. 59 (2013) 824-832. DOI:10.1373/clinchem.2012.193938 |
| [52] |
J. Xiong, H.P. Jiang, C.Y. Peng, et al., Clin. Epigenet. 7 (2015) 72. 10.1186/s13148-015-0109-x
|
| [53] |
B.F. Yuan, Methods Mol. Biol. 1562 (2017) 33-42. |
| [54] |
Q.Y. Cheng, J. Xiong, F. Wang, B.F. Yuan, Y.Q. Feng, Chin. Chem. Lett. 29 (2018) 115-118. DOI:10.1016/j.cclet.2017.06.009 |
| [55] |
T. Suzuki, T. Suzuki, Nucleic Acids Res. 42 (2014) 7346-7357. DOI:10.1093/nar/gku390 |
| [56] |
M. Taoka, Y. Nobe, Y. Yamaki, et al., Nucleic Acids Res. 46 (2018) 9289-9298. DOI:10.1093/nar/gky811 |
| [57] |
X. Cao, P.A. Limbach, Anal. Chem. 87 (2015) 8433-8440. DOI:10.1021/acs.analchem.5b01826 |
| [58] |
M. Taoka, Y. Nobe, M. Hori, et al., Nucleic Acids Res. 43 (2015) e115. 10.1093/nar/gkv560
|
| [59] |
J.A. Kowalak, E. Bruenger, T. Hashizume, et al., Nucleic Acids Res. 24 (1996) 688-693. DOI:10.1093/nar/24.4.688 |
| [60] |
B. Felden, K. Hanawa, J.F. Atkins, et al., EMBO J. 17 (1998) 3188-3196. DOI:10.1093/emboj/17.11.3188 |
| [61] |
P.F. Crain, J.D. Alfonzo, J. Rozenski, et al., RNA 8 (2002) 752-761. DOI:10.1017/S1355838202022045 |
| [62] |
B.I. Kang, K. Miyauchi, M. Matuszewski, et al., Nucleic Acids Res. 45 (2017) 2124-2136. DOI:10.1093/nar/gkw1120 |
| [63] |
S. Douthwaite, F. Kirpekar, Methods Enzymol. 425 (2007) 1-20. DOI:10.1016/S0076-6879(07)25001-3 |
| [64] |
A.M. Giessing, F. Kirpekar, J. Proteomics 75 (2012) 3434-3449. DOI:10.1016/j.jprot.2012.01.032 |
| [65] |
K.G. Patteson, L.P. Rodicio, P.A. Limbach, Nucleic Acids Res. 29 (2001) E49. 10.1093/nar/29.10.e49
|
| [66] |
J. Mengel-Jorgensen, F. Kirpekar, Nucleic Acids Res. 30 (2002) e135. 10.1093/nar/gnf135
|
| [67] |
B. Yu, Z. Yang, J. Li, et al., Science 307 (2005) 932-935. DOI:10.1126/science.1107130 |
| [68] |
M. Taucher, K. Breuker, Angew. Chem. Int. Ed. Engl. 51 (2012) 11289-11292. DOI:10.1002/anie.201206232 |
| [69] |
H. Glasner, C. Riml, R. Micura, K. Breuker, Nucleic Acids Res. 45 (2017) 8014-8025. DOI:10.1093/nar/gkx470 |
| [70] |
Y. Ruike, Y. Imanaka, F. Sato, K. Shimizu, G. Tsujimoto, BMC Genomics 11 (2010) 137. 10.1186/1471-2164-11-137
|
| [71] |
D. Serre, B.H. Lee, A.H. Ting, Nucleic Acids Res. 38 (2010) 391-399. DOI:10.1093/nar/gkp992 |
| [72] |
T.A. Down, V.K. Rakyan, D.J. Turner, et al., Nat. Biotechnol. 26 (2008) 779-785. DOI:10.1038/nbt1414 |
| [73] |
J.H. Park, J. Park, J.K. Choi, et al., BMC Med. Genomics 4 (2011) 82.
|
| [74] |
A.B. Brinkman, F. Simmer, K. Ma, et al., Methods 52 (2010) 232-236. DOI:10.1016/j.ymeth.2010.06.012 |
| [75] |
B. Yao, Y. Li, Z. Wang, et al., Mol. Cell 71 (2018) 848-857. DOI:10.1016/j.molcel.2018.07.005 |
| [76] |
C.L. Xiao, S. Zhu, M. He, et al., Mol. Cell 71 (2018) 306-318. DOI:10.1016/j.molcel.2018.06.015 |
| [77] |
G. Ficz, M.R. Branco, S. Seisenberger, et al., Nature 473 (2011) 398-402. DOI:10.1038/nature10008 |
| [78] |
K. Williams, J. Christensen, M.T. Pedersen, et al., Nature 473 (2011) 343-348. DOI:10.1038/nature10066 |
| [79] |
M. Ko, Y. Huang, A.M. Jankowska, et al., Nature 468 (2010) 839-843. DOI:10.1038/nature09586 |
| [80] |
E.A. Raiber, D. Beraldi, G. Ficz, et al., Genome Biol. 13 (2012) R69. 10.1186/gb-2012-13-8-r69
|
| [81] |
K.D. Meyer, Y. Saletore, P. Zumbo, et al., Cell 149 (2012) 1635-1646. DOI:10.1016/j.cell.2012.05.003 |
| [82] |
D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, et al., Nature 485 (2012) 201-206. DOI:10.1038/nature11112 |
| [83] |
S. Schwartz, S.D. Agarwala, M.R. Mumbach, et al., Cell 155 (2013) 1409-1421. DOI:10.1016/j.cell.2013.10.047 |
| [84] |
B. Linder, A.V. Grozhik, A.O. Olarerin-George, et al., Nat. Methods 12 (2015) 767-772. DOI:10.1038/nmeth.3453 |
| [85] |
X. Li, X. Xiong, K. Wang, et al., Nat. Chem. Biol. 12 (2016) 311-316. DOI:10.1038/nchembio.2040 |
| [86] |
D. Dominissini, S. Nachtergaele, S. Moshitch-Moshkovitz, et al., Nature 530 (2016) 441-446. DOI:10.1038/nature16998 |
| [87] |
D. Arango, D. Sturgill, N. Alhusaini, et al., Cell 175 (2018) 1872-1886. DOI:10.1016/j.cell.2018.10.030 |
| [88] |
K.D. Hansen, W. Timp, H.C. Bravo, et al., Nat. Genet. 43 (2011) 768-775. DOI:10.1038/ng.865 |
| [89] |
M.J. Booth, M.R. Branco, G. Ficz, et al., Science 336 (2012) 934-937. DOI:10.1126/science.1220671 |
| [90] |
H. Zeng, B. He, B. Xia, et al., J. Am. Chem. Soc. 140 (2018) 13190-13194. DOI:10.1021/jacs.8b08297 |
| [91] |
G. Hayashi, K. Koyama, H. Shiota, et al., J. Am. Chem. Soc. 138 (2016) 14178-14181. DOI:10.1021/jacs.6b06428 |
| [92] |
M. Yu, G.C. Hon, K.E. Szulwach, et al., Cell 149 (2012) 1368-1380. DOI:10.1016/j.cell.2012.04.027 |
| [93] |
M.J. Booth, G. Marsico, M. Bachman, D. Beraldi, S. Balasubramanian, Nat. Chem. 6 (2014) 435-440. DOI:10.1038/nchem.1893 |
| [94] |
B. Xia, D. Han, X. Lu, et al., Nat. Methods 12 (2015) 1047-1050. DOI:10.1038/nmeth.3569 |
| [95] |
X. Lu, C.X. Song, K. Szulwach, et al., J. Am. Chem. Soc. 135 (2013) 9315-9317. DOI:10.1021/ja4044856 |
| [96] |
M. Schaefer, T. Pollex, K. Hanna, F. Lyko, Nucleic Acids Res. 37 (2009) e12. 10.1093/nar/gkn954
|
| [97] |
J.E. Squires, H.R. Patel, M. Nousch, et al., Nucleic Acids Res. 40 (2012) 5023-5033. DOI:10.1093/nar/gks144 |
| [98] |
S. Edelheit, S. Schwartz, M.R. Mumbach, O. Wurtzel, R. Sorek, PLoS Genet. 9 (2013) e1003602. 10.1371/journal.pgen.1003602
|
| [99] |
U. Birkedal, M. Christensen-Dalsgaard, N. Krogh, et al., Angew. Chem. Int. Ed. Engl. 54 (2015) 451-455. |
| [100] |
N. Krogh, M.D. Jansson, S.J. Hafner, et al., Nucleic Acids Res. 44 (2016) 7884-7895. DOI:10.1093/nar/gkw482 |
| [101] |
Q. Dai, S. Moshitch-Moshkovitz, D. Han, et al., Nat. Methods 14 (2017) 695-698. DOI:10.1038/nmeth.4294 |
| [102] |
C. Riml, T. Amort, D. Rieder, et al., Angew. Chem. Int. Ed. Engl. 56 (2017) 13479-13483. DOI:10.1002/anie.201707465 |
| [103] |
A.L. Yablonovitch, P. Deng, D. Jacobson, J.B. Li, PLoS Genet.13 (2017) e1007064. 10.1371/journal.pgen.1007064
|
| [104] |
M. Sakurai, T. Yano, H. Kawabata, H. Ueda, T. Suzuki, Nat. Chem. Biol. 6 (2010) 733-740. DOI:10.1038/nchembio.434 |
| [105] |
J.M. Thomas, C.A. Briney, K.D. Nance, et al., J. Am. Chem. Soc. 140 (2018) 12667-12670. DOI:10.1021/jacs.8b06636 |
| [106] |
X. Li, P. Zhu, S. Ma, et al., Nat. Chem. Biol. 11 (2015) 592-597. DOI:10.1038/nchembio.1836 |
| [107] |
V.A. Herzog, B. Reichholf, T. Neumann, etal., Nat.Methods 14 (2017) 1198-1204. DOI:10.1038/nmeth.4435 |
| [108] |
Q.Y. Li, N.B. Xie, J. Xiong, B.F. Yuan, Y.Q. Feng, Anal. Chem. 90 (2018) 14622-14628. DOI:10.1021/acs.analchem.8b04833 |
| [109] |
E.K. Schutsky, J.E. DeNizio, P. Hu, et al., Nat. Biotechnol. 36 (2018) 1083-1090. DOI:10.1038/nbt.4204 |
| [110] |
S.U. Siriwardena, K. Chen, A.S. Bhagwat, Chem. Rev. 116 (2016) 12688-12710. DOI:10.1021/acs.chemrev.6b00296 |
| [111] |
A.E. Cozen, E. Quartley, A.D. Holmes, et al., Nat. Methods 12 (2015) 879-884. DOI:10.1038/nmeth.3508 |
| [112] |
T. Hong, Y. Yuan, Z. Chen, et al., J. Am. Chem. Soc. 140 (2018) 5886-5889. DOI:10.1021/jacs.7b13633 |
| [113] |
J. Aschenbrenner, S. Werner, V. Marchand, et al., Angew. Chem. Int. Ed. Engl. 57 (2018) 417-421. DOI:10.1002/anie.201710209 |
| [114] |
D.P. Morse, B.L. Bass, Biochemistry 36 (1997) 8429-8434. DOI:10.1021/bi9709607 |
| [115] |
X. Zhao, Y.T. Yu, RNA 10 (2004) 996-1002. DOI:10.1261/rna.7110804 |
| [116] |
Y.T. Yu, M.D. Shu, J.A. Steitz, RNA 3 (1997) 324-331. |
| [117] |
N. Liu, M. Parisien, Q. Dai, et al., RNA 19 (2013) 1848-1856. DOI:10.1261/rna.041178.113 |
| [118] |
P. Ryvkin, Y.Y. Leung, I.M. Silverman, et al., RNA 19 (2013) 1684-1692. DOI:10.1261/rna.036806.112 |
| [119] |
C. Gustafsson, B.C. Persson, J. Bacteriol. 180 (1998) 359-365. |
| [120] |
D. Lafontaine, J. Vandenhaute, D. Tollervey, Gene Dev. 9 (1995) 2470-2481. DOI:10.1101/gad.9.20.2470 |
| [121] |
Z.W. Dong, P. Shao, L.T. Diao, et al., Nucleic Acids Res. 40 (2012) e157.
|
| [122] |
A. Bakin, J. Ofengand, Biochemistry 32 (1993) 9754-9762. DOI:10.1021/bi00088a030 |
| [123] |
B.A. Flusberg, D.R. Webster, J.H. Lee, et al., Nat. Methods 7 (2010) 461-465. DOI:10.1038/nmeth.1459 |
| [124] |
C. Zhou, C. Wang, H. Liu, et al., Nat. Plants 4 (2018) 554-563. DOI:10.1038/s41477-018-0214-x |
| [125] |
Z. Liang, L. Shen, X. Cui, et al., Dev. Cell 45 (2018) 406-416. DOI:10.1016/j.devcel.2018.03.012 |
| [126] |
C.X. Song, T.A. Clark, X.Y. Lu, et al., Nat. Methods 9 (2012) 75-77. DOI:10.1038/nmeth.1779 |
| [127] |
I.D. Vilfan, Y.C. Tsai, T.A. Clark, et al., J. Nanobiotechnol. 11 (2013) 8.
|
| [128] |
B.M. Venkatesan, R. Bashir, Nat. Nanotechnol. 6 (2011) 615-624. DOI:10.1038/nnano.2011.129 |
| [129] |
J. Clarke, H.C. Wu, L. Jayasinghe, et al., Nat. Nanotechnol. 4 (2009) 265-270. DOI:10.1038/nnano.2009.12 |
| [130] |
U. Mirsaidov, W. Timp, X. Zou, et al., Biophys. J. 96 (2009) L32-L34. 10.1016/j.bpj.2008.12.3760
|
| [131] |
M. Wanunu, D. Cohen-Karni, R.R. Johnson, et al., J. Am. Chem. Soc. 133 (2011) 486-492. DOI:10.1021/ja107836t |
| [132] |
X. Lu, C. He, ChemBioChem 14 (2013) 1289-1290. DOI:10.1002/cbic.201300342 |
| [133] |
W.W. Li, L. Gong, H. Bayley, Angew. Chem. Int. Ed. Engl. 52 (2013) 4350-4355. DOI:10.1002/anie.201300413 |
| [134] |
M. Ayub, H. Bayley, Nano Lett. 12 (2012) 5637-5643. DOI:10.1021/nl3027873 |
| [135] |
D. Elmlund, H. Elmlund, Annu. Rev. Biochem. 84 (2015) 499-517. DOI:10.1146/annurev-biochem-060614-034226 |
| [136] |
S.K. Natchiar, A.G. Myasnikov, H. Kratzat, I. Hazemann, B.P. Klaholz, Nature 551 (2017) 472-477. DOI:10.1038/nature24482 |
| [137] |
F. Su, L. Wang, Y. Sun, et al., Chem. Commun. (Camb.) 51 (2015) 3371-3374. DOI:10.1039/C4CC07688E |
| [138] |
Z.Y. Wang, L.J. Wang, Q. Zhang, B. Tang, C.Y. Zhang, Chem. Sci. 9 (2018) 1330-1338. DOI:10.1039/C7SC04813K |