 2019, Vol. 30
2019, Vol. 30 


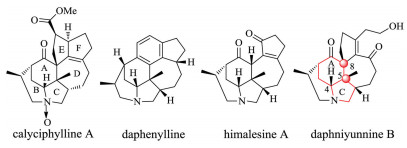
Daphniphyllum alkaloids are a class of triterpene alkaloids with complex polycyclic skeletons and interesting bioactivities. Up to now more than 320 members have been isolated and characterized from nature. According to their polycyclic skeletons, daphniphyllum alkaloids can be divided into more than 20 subgroup and calyciphylline A type may be the most complex one [1]. As shown in Fig. 1, calyciphylline A type daphniphyllum alkaloids share a unique compact 6, 6, 5, 7, 5-pentacyclic skeleton with 5-8 chiral centers among which two are challenging vicinal all carbon quaternary stereocenters [2]. They have stimulated continuous interest in the synthetic community and several elegant total syntheses have been reported [3, 4]. Li and coworkers disclosed the first total synthesis of daphenylline in 2013. In their elegant synthetic work, a gold catalyzed alkyne cyclization followed by an intramolecular Michael addition process was used to construct the bridged 6, 6, 5-tricyclic motif and a photo induced olefin isomerization/6π electrocyclization cascade followed by an oxidative aromatization process was utilized to construct the unique poly-substituted phenyl ring [4a]. And based on the optimized route to prepare the common 6, 6, 5, 7- tetracyclic intermediate in large scale, they accomplished the total syntheses of several other members of calyciphylline A type alkaloids including longeracinphyllin A [4c], daphnipaxianine A, himalenine D [4f], hybridaphniphylline B, daphnilongeranin B, daphniyunnine E and dehydrodaphnilongeranin B [4g] in the following years. Yokoshima and Fukuyama group took advantage of the characteristic conformations of 5, 6, 7-tricyclic intermediates to assemble the essential chiral side chain and they also accomplished the total synthesis of daphenylline [4b]. Based on a novel catalytic, enantioselective prototropic shift/furan Diels– Alder (IMDAF) cascade reaction to construct the 5, 6, 7-tricyclic core, Paton and Dixon group reported an elegant total synthesis of (-)-himalensine A [4d]. Zhai and co-workers completed the first total synthesis of (-)-daphnilongeranin B and a bioinspired total synthesis of (-)-daphenylline in which an intermolecular Lu's [3 + 2]cycloaddition reaction was employed to construct the challenging spiral cyclic system [4e]. Qiu group accomplished a 19-steps synthesis of daphenylline enabled by a stereoselective Mg (ClO4)2-catalyzed intramolecular amide addition cyclization and an intramolecular Diels–Alder reaction to construct its 6, 6, 5, 7- tetracyclic core architecture [4h]. Xu and coworkers disclosed a 14-steps total synthesis of (-)-himalensine A in which an intramolecular Heck reaction was used to construct the challenging 2-azabicyclo[3.3.1]nonane motif [4i]. Gao et al. developed a unified strategy to construct the 6, 6, 5, 7-tetracyclic core framework of calyciphylline A alkaloids which involved a Pd-catalyzed enolate alkenylation and a ring closing metathesis (RCM) reaction and they also completed the total synthesis of himalensine A [4j]. So it is no doubt that calyciphylline A type daphniphyllum alkaloids have served and continues to be a good platform to develop and test new synthetic methods and strategies. Inspired by all the above elegant synthetic works and continuing with our long-term research interest in synthesis of bioactive natural products [5], we recently initiated a synthetic program towards daphniyunnine B (Fig. 1), a representive member of calyciphylline A type of daphniphyllum alkaloids [6].

|
Download:
|
| Fig. 1. Structures of calyciphylline A type daphniphyllum alkaloids. | |
{kind=link}
Herein, we reported a concise synthesis of its AC bicyclic skeleton with the required 3 stereocenters (C4, C5, C8) which featured two Claisen-type rearrangement reactions to construct the required vicinal all carbon quaternary stereocenters and an intramolecular iodo-cyclization reaction to assemble the cis-confused bicyclic lactam.
In our synthetic plan, daphniyunnine B could be obtained from tricyclic ketone 1 by two lateral cyclization reactions and several oxidation state adjustment manipulations (Scheme 1). As for compound 1, it could be approached via an intramolecular α- alkylation of amide 2. The C-8 quaternary center of 2 was envisioned to be assembled by a Claisen rearrangement reaction of O-allylation product of 1, 3-diketone 3 which was expected to be obtained from bicyclic lactam 4 via an aldol coupling reaction.

|
Download:
|
| Scheme 1. Retrosynthetic analysis. | |
{kind=link}
Our synthesis started with acid-promoted Johnson-Claisen rearrangement of the known cyclohexenol (±)-5. As Scheme 2 depicted, the three-step process (rearrangement, basic hydrolysis and amidation) afforded primary amide (±)-6 in 43% overall yields. The above three steps could be performed on 10 gram scale and only one column chromatography purification was needed. Then an intramolecular iodo-cyclization under Levorse's conditions [7] followed by a DBU-promoted elimination reaction smoothly gave bicyclic lactam (±)-7 in 65% overall yields on 5 g scale. N-Protection afforded compound (±)-8 in 90% yield which NOE experiment further confirmed the cis-configuration of fused bicyclic lactam (see Supporting information for details). Subsequent SeO2-assisted allylic oxidation and Swern oxidation of the obtained diastereomeric secondary alcohols gave the key bicyclic lactam (±)-4 in high yield.

|
Download:
|
| Scheme 2. Preparation of bicyclic lactam 4. | |
{kind=link}
With the key bicyclic lactam (±)-4 in hand, we turned our attention to the assembling of the C-8 quaternary stereocenter (Scheme 3). A classic aldol reaction was utilized to combine (±)-4 and aldehyde 9. The obtained β-hydroxyl ketone (±)-10 was further transformed into more active 1, 3-diketone (±)-11 (existed as a mixture of (±)-11 and its enolization isomer (±)-12) in 80% yield. Then compounds (±)-11 and (±)-12 were submitted to an allylation reaction to give a mixture of O-alkylation product (±)-13 and C-alkylation product (±)-14 in 2.7:1 ratio and 95% total yields which could be easily seperated by column chromotography on silica gel. To our delight, the relative stereochemistry between C5 and C8 of the minor product (±)-14 was determined to be the required one by a comprehensive NOE experiment (see Supporting information for details). And the majar product 13 could also be transformed into C-alkylation products easily via a heat-promoted Claisen rearrangement reaction affording (±)-14 in 70% yield.

|
Download:
|
| Scheme 3. Synthesis of bicyclic compound 14. | |
{kind=link}
In summary, the compact AC bicyclic skeleton of daphniyunnine B with the required three stereocenters has been accomplished in 12 steps from known cyclohexenol (±)-5. Our synthesis featured two highly effective Claisen-type rearrangement reactions to assemble the requisite two vicinal all carbon quaternary stereocenters (C5 and C8) and an intramolecular iodo-cyclization reaction to assemble the cis-confused bicyclic lactam. Further synthetic works towards daphniyunine B is undertook in this laboratory now and will be reported in due course.
AcknowledgmentWe wish to thank the generous financial support from the National Natural Science Foundation of China (Nos. 21102062, 21472079).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.049.
| [1] |
(a) J. Kobayashi, T. Kubota, Nat. Prod. Rep. 26 (2009) 936-962; (b) H.F. Wu, X.P. Zhang, L.S. Ding, et al., Planta Med. 79 (2013) 1589-1598l; (C) A.K. Chattopadhyay, S. Hanessian, Chem. Rev. 117 (2017) 4104-4146. |
| [2] |
B. Kang, P. Jakubec, D. Dixon, J. Nat. Prod. Rep. 31 (2014) 550-562. DOI:10.1039/C3NP70115H |
| [3] |
(a) D. Sole, X. Urbaneja, J. Bonjoch, Org. Lett. 7 (2005) 5461-5464; (b) F. Diaba, A. Martinez-Laporta, G. Coussanes, I. Fernandez, J. Bonjoch, Tetrahedron 71 (2015) 3642-3651; (c) G. Coussanes, J. Bonjoch, Org. Lett. 19 (2017) 878-881; (d) F. Sladojevich, I.N. Michaelides, D. Benjamin, J.W. Ward, D.J. Dixon, Org. Lett. 13 (2011) 5132-5135; (e) C. Xu, L. Wang, X. Hao, D.Z.G. Wang, J. Org. Chem. 77 (2012) 6307-6313; (f) V. Wang, C. Xu, L. Chen, X. Hao, D.Z.G. Wang, Org. Lett. 16 (2014) 1076-1079; (g) M. Yang, L. Wang, Z.H. He, et al., Org. Lett. 14 (2012) 5114-5117; (h) Y. Yao, G. Liang, Org. Lett. 14 (2012) 5499-5501; (i) A.A. Ibrahim, A.N. Golonka, A.M. Lopez, J.L. Stockdill, Org. Lett. 16 (2014) 1072-1075; (j) D. Ma, H. Cheng, C. Huang, L. Xu, Tetrahedron Lett. 56 (2015) 2492-2495; (k) H. Shao, W. Bao, Z.R. Jing, et al., Org. Lett. 19 (2017) 4648-4651. |
| [4] |
(a) Z.Y. Lu, Y. Li, J. Deng, A. Li, Nat. Chem. 5 (2013) 679-684; (b) R. Yamada, Y. Adachi, S. Yokoshima, T. Fukuyama, Angew. Chem. Int. Ed. 55 (2016) 6067-6070; (c) J. Li, W.H. Zhang, F. Zhang, Y. Chen, A. Li, J. Am. Chem. Soc. 139 (2017) 14893-14896; (d) H. Shi, I.N. Michaelides, B. Darses, et al., J. Am. Chem. Soc. 139 (2017) 17755-17758; (e) X.M. Chen, H.J. Zhang, X.K. Yang, et al., Angew. Chem. Int. Ed. 57 (2018) 947-951; (f) Y.H. Chen, W.H. Zhang, L. Ren, J. Li, A. Li, Angew. Chem. Int. Ed. 57 (2018) 952-956; (g) W.H. Zhang, M. Ding, J. Li, et al., J. Am. Chem. Soc. 140 (2018) 4227-4231; (h) B. Xu, B.Y. Wang, W. Xun, F.Y.G. Qiu, Angew. Chem. Int. Ed. 58 (2019) 5754-5757; (i) Y.Y. Chen, J.P. Hu, L.D. Guo, et al., Angew. Chem. Int. Ed. 58 (2019) 7390-7394; (j) J.X. Zhong, K.W. Chen, Y.Y. Qiu, H.B. He, S.H. Gao, Org. Lett. 21 (2019) 3741-3745. |
| [5] |
(a) Z.M. Wang, Z.M. Xing, L. Liu, et al., ChemistrySelect 1 (2016) 2225-2227; (b) H. Zhang, S.Q. Ma, Z.M. Xing, et al., Org. Chem. Front. 4 (2017) 2211-2215; (c) L. Liu, H.Y. Song, P. Chen, et al., Org. Chem. Front. 5 (2018) 3013-3017; (d) T. Li, G.M. Wu, S.B. Feng, et al., Tetrahedron 75 (2019), doi: http://dx.doi.org/10.1016/j.tet.2019.06.017. |
| [6] |
(a) H. Zhang, S.P. Yang, C.Q. Fan, J. Ding, J.M. Yue, J. Nat. Prod. 69 (2006) 553-557; (b) Y.T. Di, H.P. He, Y. Lu, et al., J. Nat. Prod. 69 (2006) 1074-1076. |
| [7] |
K. Spencer, T.L. Anthony, J. Org. Chem. 53 (1988) 4006-4014. DOI:10.1021/jo00252a024 |