 2019, Vol. 30
2019, Vol. 30 


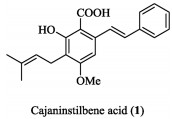
Cajaninstilbene acid (CSA, 1, Fig. 1), isolated from pigeonpea leaves [1], exhibits a wide range of pharmacological properties, including antiviral [2], anti-inflammatory [3, 4], antitumor [5, 6], anti-oxidant [7] and antibacterial [8] activity. Previously, our group reported the synthesis of a series of CSA derivatives with potent antibacterial activity against gram-positive bacteria including a "superbug" MRSA [9]. CSA was thought of as a new antibacterial chemical whose activity is different from that of other antibacterial agents.

|
Download:
|
| Fig. 1. Structure of cajaninstilbene acid. | |
CSA was first synthesized by Li's group [10] in nine steps with an overall yield of 10%, and subsequently, several synthetic strategies for synthesis of CSA were reported [11-13]. The earlier synthesis of CSA was focused on the construction of the stilbene skeleton, for which several methods have been reported [13]. However, the real difficulty in the synthesis of CSA is what has come to be known as the unsatisfactory C-prenylation reaction. The former C-prenylation reaction shown in Scheme 1 [13] used a substituted phenol 2 to couple with 3-methylbut-2-en-1-yl bromide (prenyl bromide) in a non-polar solvent. However, methods of this sort are deficient and produce an unsatisfactory yield, and isolation of the product is difficult because its polarity differs little from that of the by-products and the raw materials. In view of this situation, we adopteda strategyof pre-prenylation, i.e., before the construction of the benzyl ring to resolve this problem. For this purpose, a TiCl4-mediated [3 + 3] cyclocondensation was employed as the key reaction. Previously, Langer et al. reported the synthesis of substituted salicylates by formal [3 + 3] cyclization of 1, 3-bis (silyloxy)-1, 3-butadienes with ketene [14-16]. Using this reaction, we were able to construct the prenylated salicylate as a building block, thus achieving the pre-prenylation strategy for the synthesis of CSA. Hereinwe report our efforts on the study of a facile synthesis of CSA and its derivatives with high yields and shortroutes through a building block.

|
Download:
|
| Scheme 1. The earlier C-prenylation reaction. | |
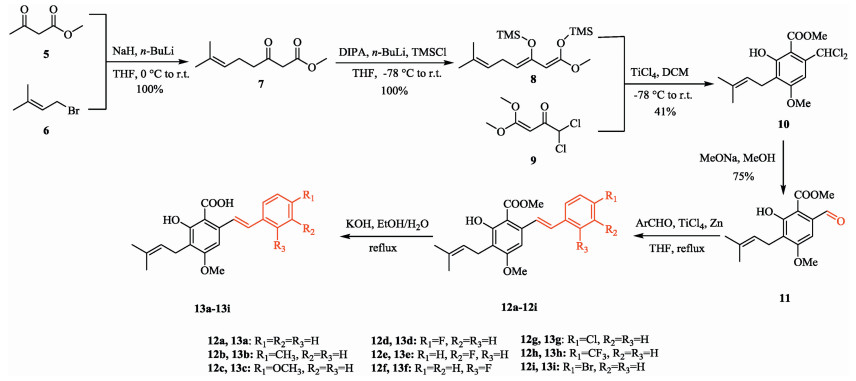
Our synthesis began with the prenylation reaction of methyl acetoacetate with 3-methylbut-2-en-1-yl bromide shown in Scheme 2. Compound 7 was prepared by this reaction according to a published method [17] and was then treated with newly prepared LDA and TMSCl to obtain prenylated 1, 3-bis(silyloxy)-1, 3-butadiene 8. This product is unstable in air and was therefore used in subsequent steps without any further purification. Compound 9 was prepared according to an established method [18], but the dichloroacetic anhydride was replaced by the much cheaper reagent dichloroacetyl chloride, which gives an acceptable yield. Subsequently, a TiCl4-mediated [3 + 3] cyclization of 8 with compound 9 was performed to afford the substituted salicylate 10 in a moderate yield. This compound was then treated with sodium methoxide in methanol and subsequently acidified with hydrochloric acid to obtain the building block methyl 6-formyl-2- hydroxy-4-methoxy-3-(3-methylbut-2-en-1-yl) benzoate 11 [18].

|
Download:
|
| Scheme 2. The facile synthesis of cajaninstilbene acid and its derivatives. | |
With compound 11 in hand, efforts were made to introduce the styryl group. Initially, a Wittig reaction was attempted but when compound 11 was treated with triphenyl phosphonium ylide under modified Wittig reaction conditions [19], the reaction gave a satisfactory yield but an inseparable mixture of cis- and trans-stilbenoids (Z:E = 42:58) resulted. Three ylides were tested as reagents but all gave similar results, shown in Table 1. The Wittig-Horner reaction and Julia coupling reaction both failed to give a satisfactory yield in spite of numerous trials and this appeared to be due to the presence of the phenolic hydroxyl group. Use of protective groups for this hydroxyl group will complicate the synthetic route and thus was not undertaken.
|
|
Table 1 The yield and ratio of isomers in a Wittig reaction.a |
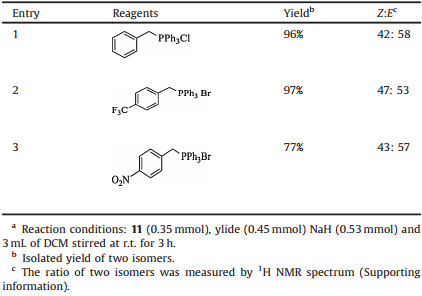
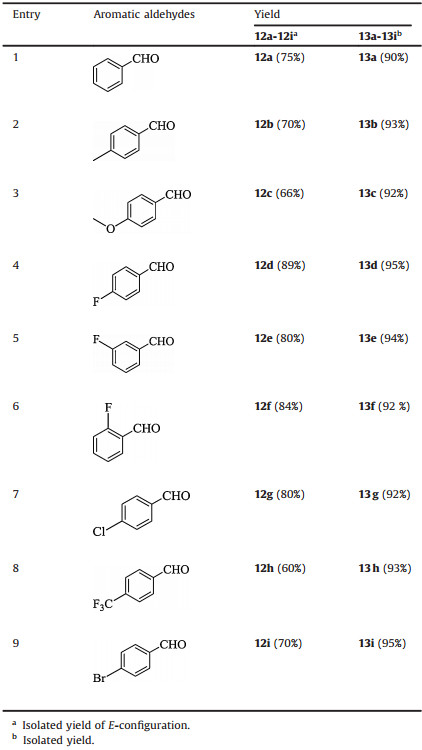
Fortunately, a low-valency titanium mediated McMurry coupling [20, 21] was found to be especially suitable at this stage and enabled compound 11 to be coupled with different aromatic aldehydes to afforded stilbenoids (12a-12i) with acceptable yields (Table 2) and high selectivity for trans-configured products that can be easily crystallized from methanol. Cajaninstilbene acid and its derivatives (13a-13i) were obtained by hydrolysis of 12a-12i. In this way, we accomplished the synthesis of cajaninstilbene acid and its derivatives through a building block in only six steps. The synthesis route is shorter than those previously reported and produces a much higher overall yield [11, 13].
|
|
Table 2 The yield for synthesis of cajaninstilbene acid and its derivatives. |
{kind=link}
{kind=link}
{kind=link}
Finally, we have completed a concise six step synthesis of cajaninstilbene acid and its derivatives with high yields. Our synthetic route is the most concise method with the higher yield for synthesis of CSA, and no protecting groups were used. Currently, we are able to obtain the CSA and its derivatives easily, which will support further medicinal chemistry studies of CSA.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (No. 81673336).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.043.
| [1] |
C.J. Cooksey, J.S. Dahiya, P.J. Garratt, et al., Phytochemistry 21 (1980) 2935-2938. |
| [2] |
X.Y. Ji, J.H. Chen, G.H. Zheng, et al., J. Med. Chem. 59 (2016) 10268-10284. |
| [3] |
M.Y. Huang, J. Lin, K. Lu, et al., J. Agric. Food. Chem. 64 (2016) 2893-2900. |
| [4] |
R. Schuster, W. Holzer, H. Doerfler, et al., Food Funct. 7 (2016) 3798-3806. |
| [5] |
Y. Fu, O. Kadioglu, B. Wiench, et al., Phytomedicine 22 (2015) 462-468. |
| [6] |
O. Kadioglu, Y. Fu, B. Wiench, et al., Arch. Toxicol. 90 (2016) 575-588. |
| [7] |
L. Liang, M. Luo, Y. Fu, et al., Toxicol. Lett. 219 (2013) 254-261. |
| [8] |
Y. Kong, Y.J. Fu, Y.G. Zu, et al., Food Chem. 121 (2010) 1150-1155. DOI:10.1016/j.foodchem.2010.01.062 |
| [9] |
Z.Z. Geng, J.J. Zhang, J. Lin, et al., Eur. J. Med. Chem. 100 (2015) 235-245. DOI:10.1016/j.ejmech.2015.06.008 |
| [10] |
X.Y. Ji, S.T. Xue, G.H. Zheng, et al., Acta Pharm. Sin. B. 1 (2011) 93-99. DOI:10.1016/j.apsb.2011.06.005 |
| [11] |
G.S. Grandhi, J. Selvakumar, S. Dana, et al., J. Org. Chem. 83 (2018) 12327-12333. |
| [12] |
I.S. Aidhen, R. Mukkamala, C. Weidner, et al., Org. Lett. 17 (2015) 194-197. |
| [13] |
X.J. Xu, T. Zeng, Z.X. Huang, et al., J. Nat. Prod. 81 (2018) 2621-2629. |
| [14] |
C. Mamat, T. Pundt, T. Dang, et al., Eur. J. Org. Chem. 3 (2008) 492-502. |
| [15] |
S. Reimann, A. Bunescu, R. Litschko, J. Fluor. Chem. 139 (2012) 28-45. |
| [16] |
A. Bunescu, S. Reimann, M. Lubbe, et al., J. Org. Chem 74 (2009) 5002-5010. |
| [17] |
J.E. Resek, J. Org. Chem. 73 (2008) 9792-9794. |
| [18] |
V. Karapetyan, S. Mkrtchyan, G. Ghazaryan, et al., Tetrahedron 65 (2009) 9271-9279. |
| [19] |
T. Ismail, S. Shafi, J. Srinivas, et al., Bioorg. Chem. 64 (2016) 97-102. |
| [20] |
J.E. McMurry, M.P. Fleming, J. Am. Chem. Soc. 96 (1976) 4708-4709. |
| [21] |
R. Baskin, M. Gali, S.O. Park, et al., Bioorg. Med. Chem. Lett. 22 (2012) 1402-1407. |