 2019, Vol. 30
2019, Vol. 30 


b Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong, China
Heterocycles feature prominently in many molecules of importance in biological, agricultural and medicinal chemistry [1-7]. Often, more than one heteroatom-the most common of them being oxygen and/or nitrogen-are found in such heterocycles. The substitution pattern, ring size, and stereochemistry of heterocyclic moieties in these compounds are varied, and hence new synthetic strategies, particularly those based on green starting materials and methods, are desirable for obtaining these heterocycles.
We have been studying both the inter- (Scheme 1) and intramolecular (4 + 3) cycloadditions (Scheme 2) of epoxy enolsilanes with furan that proceeded under silyl triflate catalysis [8]. While the intermolecular cycloaddition proceeded without diastereoselectivity to generate both the endo 2 and the exo 3 cycloadducts, the intramolecular (4 + 3) cycloaddition of 4 was highly endo selective to provide polycylic adduct 5. The oxabicyclic or oxatricyclic adducts 2, 3, 5 have been obtained with highly conserved ee from enantiomerically enriched epoxides [9], and this has been attributed to the stereochemically-defined, activated epoxides, rather than the achiral oxyallyl cations, that are the electrophilic species undergoing cycloaddition [10].

|
Download:
|
| Scheme 1. Intermolecular (4 + 3) cycloadditions of an epoxy enolsilane with furan. | |

|
Download:
|
| Scheme 2. Intramolecular (4 + 3) cycloadditions of an epoxy enolsilane with furan. | |
To further explore the scope of the (4 + 3) cycloadditions, we aimed to explore intramolecular cycloaddition substrates which bear a heteroatom-containing tether, to install additional heteroatomic features for constructing more complex heterocyclic frameworks. We also noted that these heteroatom-tethered cycloaddition substrates 8a-c can be obtained from furfural 7, a compound currently being produced at about 280 kTon annually from lignocellulosic biomass [11], making this strategy a more green synthesis of hetero-polycycles. As a proof of concept, we explored the synthesis of several nitrogen-tethered furan substrates, and examined their cycloadditions for the green synthesis of bis-heteroatomic tricyclic adducts.
To explore the feasibility of a synthesis of these nitrogencontaining heterocycles from a (4 + 3) cycloaddition strategy, substrates 8a-c were designed that join the furan to the epoxy enolsilane moiety by a tether bearing nitrogen. Cycloaddition substrates 8a-c were synthesized as shown in Scheme 3 from furfural 7, an abundant starting material. Reductive amination with allylamine afforded amine 9 [12], which was N-protected using three different protecting groups to give allylic amines 10a-c. Each of these were homologated by cross-metathesis with methyl vinyl ketone to provide 11a-c. Racemic epoxy ketones 12a-c were obtained by nucleophilic epoxidation. Asymmetric epoxidation under Deng's conditions [13] provided epoxyketone (-)-12a in 83% ee after recrystallization. Deprotonation and silylation afforded cycloaddition substrates 8a-c (for details refer to the Supporting Information).

|
Download:
|
| Scheme 3. Synthesis of cycloaddition substrates 8a-c. | |
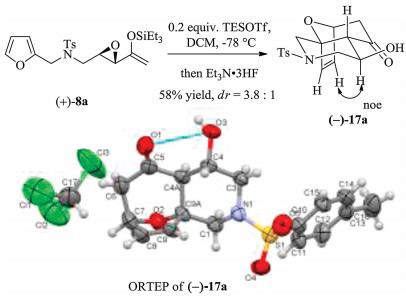
Under the typical reaction conditions using a catalytic amount of TESOTf at low temperatures, (4 + 3) cycloaddition of 8a proceeded, and provided cycloadducts 17a and 18a bearing piperidine-fused frameworks, with 17a being the major diastereomer (Table 1, entry 2, for details refer to the Supporting Information). The structures and relative stereochemistries of cycloadducts 17a and 18a were determined to be derived from endo and exo cycloadditions respectively, deduced based on NOE experiments as shown in Scheme 4.

|
Download:
|
| Scheme 4. Intramolecular (4 + 3) cycloaddition of epoxy enolsilane (+)-8a. | |
Moreover, the (4 + 3) cycloaddition of (+)-8a of 83% ee generated (-)-17a and (+)-18a of 86% ee and 84% ee, respectively. The X-ray crystallographic analysis of (-)-17a confirmed its absolute stereochemistry to be as shown (Flack parameter = 0.11). These results showed that the intramolecular cycloaddition also proceeded, not by an oxyallylic cation, but via the intermediacy of an activated epoxide that directed backside attack and whose absolute stereochemistry directed the cycloaddition.
At the start, it may be presumed that the introduction of a heteroatom into the tether of an intramolecular (4 + 3) cycloaddition substrate would not lead to any remarkable differences in the reactivity or the outcome of the cycloaddition reaction, yet this was not the case. Although 8a reacted under similar conditions as in the cycloaddition of 4 having an all-carbon tether (Table 1, entries 1 and 2), the cycloaddition of substrate 8b bearing a less electronwithdrawing carbamate protecting group was largely incomplete using a catalytic amount of TESOTf (Table 1, entry 3). A large amount of the desilylated substrate 12b was recovered after workup. Ultimately, a stoichiometric amount of TESOTf was required for full conversion (Table 1, entry 4). We surmised that the Lewis basic heteroatoms in the tether of 8b could be sequestering the silyl triflate, making it less available for inducing cycloaddition. Using TfOH as a catalyst [14] did not improve the cycloaddition results (Table 1, entry 5). Observations were similar for the CBz-protected substrate 8c (Table 1, entry 6).
|
|
Table 1 Intramolecular (4 + 3) cycloadditions of epoxy enolsilanes with furans. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
In addition, diminished cycloaddition yields were observed in the reactions of 8b and 8c compared with 8a: this was because both reactions were further complicated by the generation of cyclic carbamate side product 19, which was isolated in 5% and 12% yields respectively. This side product was deduced to arise, as shown in Scheme 5, from the attack of the activated epoxide by the carbamate protecting group that consumed some of the starting materials 8b and 8c.

|
Download:
|
| Scheme 5. Proposed mechanism of the formation of side product 19. | |
{kind=link}
The diastereoselectivities of these cycloadditions were also lower than expected. In contrast to the exclusive endo-selectivity in the cycloaddition of 4 (Table 1, entry 1), the reaction of 8a generated endo- and exo-diastereomers 17a and 18a with dr = 4.0:1. The reactions of 8b and 8c produced piperidine-fused heterocyclic adducts 17b/c and 18b/c, but the endo/exo selectivities were even lower (Table 1, entries 4, 6).
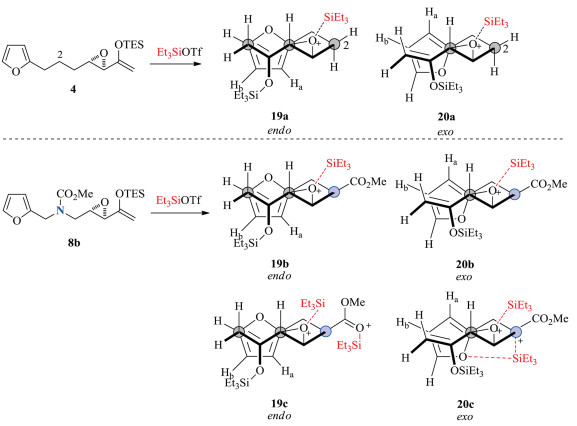
The stereoselective synthesis of bis-heteroatomic polycyclic adducts using this (4 + 3) cycloaddition route was more challenging than expected. The presence of an amine group in the tethers of the intramolecular (4 + 3) cycloaddition substrates was not initially anticipated to induce significant effects on the cycloaddition compared with the reaction of 4, as computational and experimental studies on an intramolecular Diels-Alder reaction showed that the effects of a heteroatom-containing tether compared with an all-carbon linkage was negligible [15]. From the start, the heteroatom tether necessitated reaction conditions with a higher concentration of Lewis acid. We surmise that the heteroatom effect was not likely to be steric in origin. In fact, transition state 19a resulting in the endo cycloadduct has typically been the more favoured, as there is better alignment of the atoms undergoing cycloaddition compared with 20a (Scheme 6). It is not obvious how heteroatoms replacing the carbon at the C2 position, as typified by substrate 8b in Scheme 6, would significantly sterically alter the stability of 19b relative to 20b. We conjecture that the effect may be electronic, and one possibility may be that additional association of the protected amine group with the silylating agent could lead to a pre-organization of substrate 8b which favoured 20c. However, computations will be needed to elucidate with more certainty the factors at play.

|
Download:
|
| Scheme 6. Proposed cycloaddition models of endo/exo diastereoselectivity. | |
{kind=link}
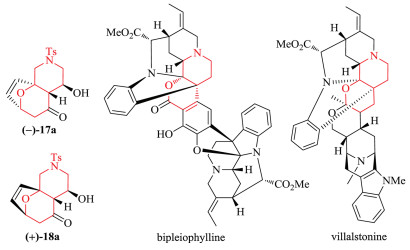
In summary, complex bis-heteroatomic polycyclic adducts were synthesized from the (4 + 3) cycloadditions of epoxy enolsilanes bearing N atoms in the tether. Amino-oxacycle 17a (Fig. 1) was obtained with 4:1 diastereoselectivity in a good yield, and with up to 86% ee. The minor exo cycloadduct 18a was also obtained with 84% ee. The optical purity of 17a can be improved by increasing the enantioselectivity of the asymmetric epoxidation. The aminooxacycle motif is found in natural products like bipleiophylline [16] and villalstonine [17] (Fig. 1). Since the cycloaddition substrates can be readily obtained from furfural, this constitutes a relatively green synthesis for such heterocycles. Other bis-heteroatomic adducts 17 and 18 have also been synthesized, and in theory can be obtained in optically enriched form as demonstrated for 17a. However, generally the (4 + 3) cycloadditions of substrates 8b-c underwent less diastereoselective (4 + 3) cycloadditions, wherein the protected amine group in the tether influenced the cycloaddition rather significantly.

|
Download:
|
| Fig. 1. Structures of natural products with amino-oxacycle motifs. | |
{kind=link}
Acknowledgments
This work was supported by the Research Grants Council General Research Fund (No.17301314); the State Key Laboratory of Synthetic Chemistry; and the University of Hong Kong. We thank the University of Hong Kong for a University Postgraduate Fellowship to JH, a Hung Hing Ying Postgraduate Scholarship to JPLN, and exploratory work by Dr. Kong Ching Wong.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.051.
| [1] |
T.Y. Zhang, The evolving landscape of heterocycles in drugs and drug candidates, in: E.F.V. Scriven, C.A. Ramsden (Eds.), Heterocyclic Chemistry in the 21st Century, Advances in Heterocyclic Chemistry, Vol.121, Elsevier, New York, 2017, pp. 1-12. https://www.sciencedirect.com/science/article/pii/S0065272516300460
|
| [2] |
C.M. Marson, Saturated heterocycles with applications in medicinal chemistry, in: E.F.V. Scriven, C.A. Ramsden (Eds.), Heterocyclic Chemistry in the 21st Century, Advances in Heterocyclic Chemistry, Vol. 121, Elsevier, New York, 2017, pp. 13-33. https://www.sciencedirect.com/science/article/pii/S0065272516300198
|
| [3] |
Y.J. Wu, Heterocycles and medicine: a survey of the heterocyclic drugs approved by the US FDA from 2000 to present, in: G.W. Gribble, J.A. Joule (Eds.), Progress in Heterocyclic Chemistry, Vol. 24, Elsevier, New York, 2012, pp.1-53. https://www.sciencedirect.com/science/article/pii/B9780080968070000014
|
| [4] |
R.D. Taylor, M. MacCoss, A.D.G. Lawson, J. Med. Chem. 57 (2014) 5845-5859. DOI:10.1021/jm4017625 |
| [5] |
A.F. Pozharskii, A.T. Soldatenkov, A.R. Katritzky, Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry and Applications, 2nd edition, John Wiley & Sons, Ltd., West Sussex, 2011.
|
| [6] |
A. Gomtsyan, Chem. Heterocycl. Compd. 48 (2012) 7-10. DOI:10.1007/s10593-012-0960-z |
| [7] |
C. Lamberth, Pest Manag. Sci. 69 (2013) 1106-1114. DOI:10.1002/ps.3615 |
| [8] |
W.K. Chung, S.K. Lam, B. Lo, et al., J. Am. Chem. Soc. 131 (2009) 4556-4557. DOI:10.1021/ja807566t |
| [9] |
B. Lo, S. Lam, W.T. Wong, P. Chiu, Angew. Chem. Int. Ed. 51 (2012) 12120-12123. DOI:10.1002/anie.201207427 |
| [10] |
E.H. Krenske, S. Lam, J.P.L. Ng, et al., Angew. Chem. Int. Ed. 54 (2015) 7422-7425. DOI:10.1002/anie.201503003 |
| [11] |
R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba, M.L. Granados, Energy Environ. Sci. 9 (2016) 1144-1189. DOI:10.1039/C5EE02666K |
| [12] |
A. Palma, M. Artelsmair, G. Wu, et al., Angew. Chem. Int. Ed. 56 (2017) 15688-15692. DOI:10.1002/anie.201706487 |
| [13] |
X. Lu, Y. Liu, B. Sun, B. Cindric, L. Deng, J. Am. Chem. Soc. 130 (2008) 8134-8135. DOI:10.1021/ja802982h |
| [14] |
B. Lo, P. Chiu, Org. Lett. 13 (2011) 864-867. DOI:10.1021/ol102897d |
| [15] |
M.N. Paddon-Row, A.I. Longshaw, A.C. Willis, M.S. Sherburn, Chem.-Asian J. 4 (2009) 126-134. DOI:10.1002/asia.200800352 |
| [16] |
T.S. Kam, S.J. Tan, S.W. Ng, K. Komiyama, Org. Lett. 10 (2008) 3749-3752. DOI:10.1021/ol801354s |
| [17] |
K. Ghedira, M. Zeches-Hanrot, B. Richard, et al., Phytochemistry 27 (1988) 3955-3962. DOI:10.1016/0031-9422(88)83053-X |