 2019, Vol. 30
2019, Vol. 30 


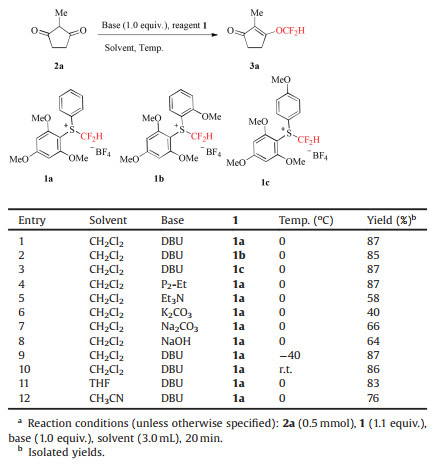
The difluoromethyl ethers represent a valuable class of organofluorine compounds, which are wide applied in pharmaceuticals [1], agrochemicals [2] and materials [3]. Particularly, the difluoromethoxy group (OCF2H) is an important structural motif in medicinal chemistry and drug discovery, since it is capable of acting as a lipophilic hydrogen-bond donor [4]. For example, pantoprazole is used for the treatment of gastrohelcoma, which is well known as an irreversible proton pump inhibitor [5], and roflumilast, a phosphodiesterase inhibitor, is employed to treat chronic obstructive pulmonary disease (COPD) [6], GRN-529 is in clinic trials stage as a candidate drug for treatment of autistic children [7] (Fig. 1). Therefore, the synthesis of organic molecules containing OCF2H group have attracted considerable attention in the past decades.

|
Download:
|
| Fig. 1. Drugs containing OCF2H building block. | |
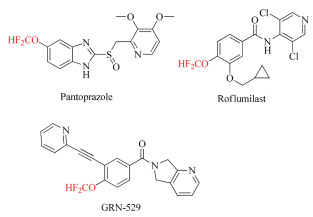
Conventionally, organic molecules bearing OCF2H moiety are prepared by O-difluoromethylation of phenols and alcohols with in situ generated difluorocarbene [8], and a lot of difluorocarbene precusors such as TMSCF2Br, ClF2CCOONa, PDFA, BrCF2CO2Et, FSO2CF2COOH, HCF2OTf, HCF3, and CF2Br2 have been developed for this purpose. However, these methodologies recorded previously mainly focus on the difluoromethylation of phenols and aliphatic alcohols to form the difluoromethyl ethers, few protocols for the synthesis of difluoromethyl enol ethers are reported. Up to date, only three groups documented the approaches for the access to difluoromethyl enol ethers (Scheme 1). Shibata and coworkers disclosed selective O-difluoromethylation of 1, 3-diones by S-(bromodifluoromethyl)diaryl sulfonium salts mediated by a strong organic base, but excess of 2.0 equiv. of substrates were required due to the nature of the reagents, although a wide range of 1, 3-diones are applicable to the reaction in good to excellent yields [9] (Scheme 1a). Weng et al. reported that ethyl chlorodifluoroacetate is a suitable reagent for the O-difluoromethylation of 1, 3-diones. However, this protocol required a high reaction temperature [10] (Scheme 1b), besides, usage of DMF as solvent also limits its applicability because of troublesome work up process. Afterwards, Xiao group described a mild process for the synthesis of difluoromethyl enol ethers from 1, 3-diones via direct transfer of the CF2H group and difluocarbene pathway, although only limited examples were demonstrated [11] (Scheme 1c). Later on, Xiao and co-workers also reported a protocol of O-difluoromethylation of 1, 3-diones using PDFA as the difluorocarbene precursor [12]. And very recently, we designed and synthesized the bench-stable S-(difluoromethyl) sulfonium salts 1 as a highly efficient electrophilic difluoromengylating reagent and difluorocarbene source [13]. As a part of our continuous research for the development of efficient methodologies for the synthesis of fluoro-organic compounds, we are herein interested in the potentiality of our reagent 1 for the difluoromethylation of 1, 3-diones to furnish the difluoromethyl enol ethers. And the results showed that a widevariety of 1, 3-dions can smoothly occur reaction to give corresponding difluoromethyl enol ethers in good to excellent yields under mild conditions (Scheme 1d).

|
Download:
|
| Scheme 1. Synthesis of difluoromethyl enol ethers from 1, 3-diones. | |
Our research commenced with the optimization of reaction conditions, 2-methyl-1, 3-cyclopentanedione 2a was chosen as a model substrate, and the results was summarized in Table 1. when 2a and 1a were treated by 1.0 equiv. of DBU in the dichloromethane at 0 ℃, exclusive O-difluorothylation pruduoct, difluoromethyl enol ether 3a, was isolated in 87% yield (entry 1), and no C-selective difluoromethylation product was detected in this case. Thus, it is very interesting in contrast to the protocol of the C-selective difluoromethylation of β-ketoesters with 1a reported recently by us [12a]. Furthermore, other reagents such as 1b and 1c also gave the comparable results under the same conditions (entries 2 and 3). Encouraged by these results, we further examined other bases, including organic and inorganic bases (entries 4–8). P2-Et also performed good to provided 3a in 87% isolated yield (entry 4), but the weak organic base Et3N led to a lower yield of 58% (entry 5). It was worth noting that organic bases were superior to inorganic bases in this reaction system, probably arising from the worse solubility of inorganic bases in dichloromethane (entries 6–8). Next, the reaction temperature was surveyed, and reaction proceeding at both -40 ℃ and ambient temperature gave the similar yields to that of conducting at 0 ℃ (entries 9 and 10), ascertaining that this process is not sensitive to reaction temperature, and possessing a wide window of temperature. Finally, the effect of solvents was investigated, usage of THF instead of dichloromethane slightly decreased yield to 83% (entry 11), and 3a was afforded in a visibly lower yield of 76% when the reaction was carried out in the acetonitrile (entry 12). Thus, dichloromethane was identified as the best choice.
|
|
Table 1 Survey of reaction conditions.a |
{kind=link}
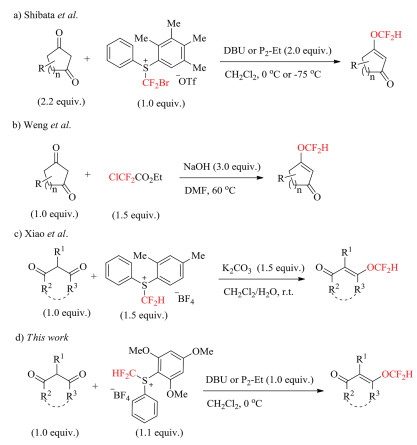{kind=link}
With the optimized reaction conditions in hand, we evaluated the capability and generality of this protocol for the O-difluoromethylation of various 1, 3-diones. As shown in Scheme 2, a broad range of 1, 3-cyclopentanediones 2a-f with different substituents at the C-2 position were applicable to this process to afford corresponding difluoromethyl enol ethers 3a-f in excellent yields. Notably, a wide variety of 1, 3-indandiones 2g-m bearing various substituents at C-2 position were also tolerated to furnish O-difluoromethylation products in good to high yields. Furthermore, 1, 3-cyclohexanediones were also the suitable substrates, and a series of 1, 3-cyclohexanediones 2n-z were smoothly transferred into corresponding difluoromethyl enol ethers 3n-z in high to excellent yields. And same to 1, 3-cylcopentanediones substrates, 1, 3-cyclohexanediones possessing diverse substituents at C-2 position, including halogen (2o), methyl (2p), benzyl (2q-v), also provided desired products 3o-v in excellent yields. It was worth noting that a variety of functional groups such as fluoro, chloro, bromo, nitrile, or nitro moieties on the aromatic rings, were tolerance in this reaction. Besides, 1, 3-cyclohexanediones having methyl, isopropyl, phenyl, and dimethyl substituents at C-5 position were all compatible with this approach to produce corresponding difluoromethyl enol ethers in high to excellent yields. Remarkably, acyclic 1, 3-dione 2aa could also undergo Odifluoromethylation under the standard reaction conditions to deliver difluoromethyl enol ethers, albeit in a lower isolated yield of 39%.

|
Download:
|
| Scheme 2. Reaction conditions (unless otherwise specified): 2 (0.5 mmol), DBU (0.5 mmol, 1.0 equiv.), 1a (0.55 mmol, 1.1 equiv.), CH2Cl2 (3.0 mL), 0 ℃, 20 min. Isolated yields. a P2-Et was used as the base. | |
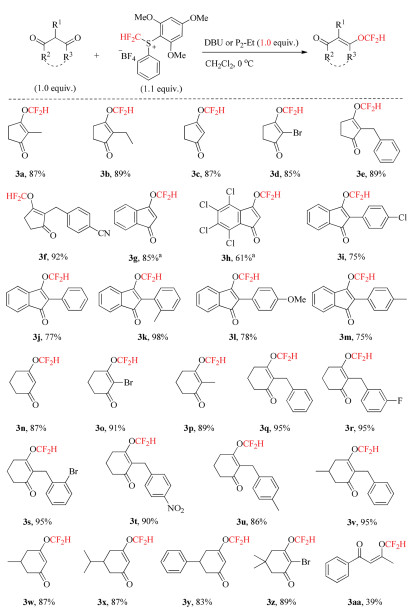{kind=link}
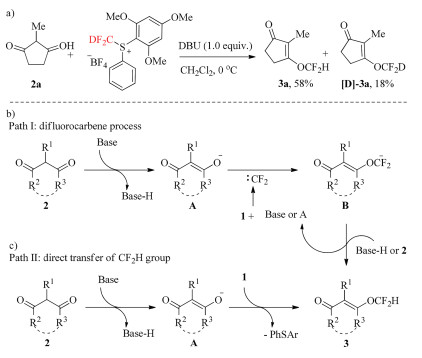
According to the mechanism proposed by other groups, the O-difluoromethylation of 1, 3-diones might occur via direct transfer of CF2H group and difluorocarbene process. In order to gain more information in our case, control experiment was carried out. The usage of deuterium-labelled reagent [D]-1a instead of 1a, deuterium-labelled product [D]-3a was obtained in 18% yield, but 58% 3a was observed (Scheme 3a), indicating that the reaction would be likely proceed involving in difluorocarbene process as the main pathway. Therefore, on the basis of the experimental data above and literatures reported previously by other groups, we propose the reaction mechanism as shown in Scheme 3c. 1, 3-Diones was treated by base to deliver enolate anion A. Subsequently, A react with difluorocarbene generated from reagent 1 and base or A to give difluoromethylated anion B, followed by protonating to form difluromomethyl enol ether (path Ⅰ, Scheme 3b). However, the direct attack of enolate anion to CF2H moiety of sulfonium salt could not be ruled out (path Ⅱ, Scheme 3c).

|
Download:
|
| Scheme 3. Control experiments and plausible reaction mechanism for the O-difluoromethylation of 1, 3-diones by S-(difluoromethyl)sulfonium salt 1. | |
{kind=link}
In summary, we have developed a facile approach for the selective O-difluoromethylation of 1, 3-diones. And a wide variety of 1, 3-diones readily reacted with bench-stable S-(difluoromethyl) sulfonium salt in the presence of organic base to furnish corresponding difluoromethyl enol ethers in good to excellent yields. Mechanistic studies revealed that the reaction proceeds mainly via the difluorocarbene process, albeit the direct transfer of CF2H group could not be ruled out.
AcknowledgmentThis work was supported by the Science and Technology Foundation of Shenzhen, Shenzhen Science and Technology Innovation Committee (No. JCYJ20170818143001461). We thank Instrumental Analysis Center of Shenzhen University (Xili Campus) for analytical work.
| [1] |
(a) B.E. Smart, Chem. Rev. 96 (1996) 1555-1556; (b) N. Chauret, D. Guay, C. Li, et al., Bioorg. Med. Chem. Lett. 12 (2002) 2149-2152; (c) T. Ohmine, T. Katsube, Y. Tsuzaki, et al., Bioorg. Med. Chem. Lett. 12 (2002) 739-742; (d) P. Jeschke, E. Baston, F.R. Leroux, Mini-Rev. Med. Chem. 7 (2007) 1027-1034 and references therein; (e) D. McKerrecher, K.G. Pike, M.J. Waring, PCT Int. Appl. WO (2007) 2007007042A1; (f) W.K. Hagmann, J. Med. Chem. 51 (2008) 4359-4369; (g) T. Furuya, C. Kuttruff, T. Ritter, Curr. Opin. Drug Discovery Dev. 11 (2008) 803-819; (h) J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432-2506; (i) M. Bassetto, S. Ferla, F. Pertusati, Future Med. Chem. 7 (2015) 527-546. |
| [2] |
(a) W.F. Goure, K.L. Leschinsky, S.J. Wratten, J.P. Chupp, J. Agric. Food Chem. 39 (1991) 981-986; (b) R.A. Pérez, C. Sánchez-Brunete, E. Miguel, J.L. Tadeo, J. Agric. Food Chem. 46 (1998) 1864-1869. |
| [3] |
(a) P. Kirsch, B. Bremer, Angew. Chem. Int. Ed. 39 (2000) 4216-4235; (b) T. Tasaka, S. Takenaka, K. Kabu, Y. Morita, H. Okamoto, Ferroelectronics 276 (2002) 83-87; (c) O.V. Boltalina, T. Nakajima, New Fluorinated Carbons: Fundamentals and Applications, Elsevier, Amsterdam, 2016. |
| [4] |
(a) J.A. Erickson, J.I. McLoughlin, J. Org. Chem. 60 (1995) 1626-1631; (b) Y. Zafrani, D. Yeffet, G. Sod-Moriah, et al., J. Med. Chem. 60 (2017) 797-804. |
| [5] |
(a) W.A. Simon, C. Büdingen, S. Fahr, B. Kinder, M. Koske, Biochem. Pharmacol. 42 (1991) 347-355 (b) B. Kohl, E. Sturm, G. Rainer, United States Patent, US 4758579 (2019). |
| [6] |
(a) L. Marin, P. Colombo, M. Bebawy, P.M. Young, D. Traini, Expert Opin. Drug Delivery 8 (2011) 1205-1220; (b) M. Cazzola, S. Picciolo, M.G. Matera, Expert Opin. Pharmacother. 11 (2010) 441-449. |
| [7] |
(a) D.Z. Li, S.V. O'Neil, D.M. Springer, B.M. Zegarelli, PCT Int. Appl (2019) WO2010124047A1; (b) L. Zhang, G. Balan, G. Barreiro, et al., J. Med. Chem. 57 (2014) 861-877; (c) J.B. Sperry, R.M. Farr, M. Levent, et al., Org. Process Res. Dev.16 (2012) 1854-1860. |
| [8] |
(a) B.R. Langlois, J. Fluorine Chem. 41 (1988) 247-261; (b) C.S. Thomoson, W.R. Dolbier, J. Org. Chem. 78 (2013) 8904-8908; (c) I. Rico, C. Wakselman, Tetrahedron Lett. 22 (1981) 323-326; (d) Q.Y. Chen, S.W. Wu, J. Fluorine Chem. 44 (1989) 433-440; (e) L. Zhang, J. Zheng, J. Hu, J. Org. Chem. 71 (2006) 9845-9848; (f) J. Zheng, Y. Li, L. Zhang, et al., Chem. Commun. (2007) 5149-5151; (g) Y. Zafrani, G. Sod-Moriah, Y. Segall, Tetrahedron 65 (2009) 5278-5283; (h) J.B. Sperry, K. Sutherland, Org. Process Res. Dev. 15 (2011) 721-725; (i) F. Wang, W. Huang, J. Hu, Chin. J. Chem. 29 (2011) 2717-2721; (j) P.S. Fier, J.F. Hartwig, Angew. Chem. Int. Ed. 52 (2013) 2092-2095; (k) A. Polley, G. Bairy, P. Das, R. Jana, Adv. Synth. Catal. 360 (2018) 4161-4167; (l) L. Li, F. Wang, C. Ni, J. Hu, Angew. Chem. Int. Ed. 52 (2013) 12390-12394; (m) X.Y. Deng, J.H. Lin, J. Zheng, J.C. Xiao, Chem. Commun. 51 (2015) 8805-8808; (n) J. Mizukado, Y. Matsukawa, H.D. Quan, M. Tamura, A. Sekiya, J. Fluorine Chem. 127 (2006) 400-404; (o) Y. Hagooly, O. Cohen, S. Rozen, Tetrahedron Lett. 50 (2009) 392-394; (p) Q. Xie, C. Ni, R. Zhang, et al., Angew. Chem. Int. Ed. 56 (2017) 3206-3210. |
| [9] |
G. Liu, X. Wang, X.H. Xu, et al., Org. Lett. 15 (2013) 1044-1047. DOI:10.1021/ol4000313 |
| [10] |
X. Lin, Z. Weng, Org. Biomol. Chem. 13 (2015) 3432-3437. DOI:10.1039/C5OB00020C |
| [11] |
C.B. Yue, J.H. Lin, J. Cai, et al., RSC Adv. 6 (2016) 35705-35708. DOI:10.1039/C6RA06338A |
| [12] |
C. Liu, X.Y. Deng, X.L. Zeng, et al., J. Fluorine Chem. 192 (2016) 27-30. DOI:10.1016/j.jfluchem.2016.10.011 |
| [13] |
(a) S.L. Lu, X. Li, W.B. Qin, et al., Org. Lett. 20 (2018) 6925-6929; (b) G.K. Liu, S.L. Lu, X. Li, W.B. Qin, PCT Int. Appl. CN (2018) 2018113547. |