 2019, Vol. 30
2019, Vol. 30 


b Department of Chemistry, The Hong Kong University of Science and Technology (HKUST), Hong Kong, China
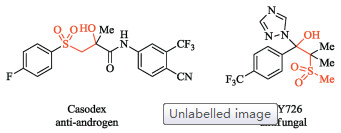
Vicinal difunctionalization of olefins, which is to add two functional groups across the carbon-carbon double bond, is a fundamentally important process in organic synthesis [1, 2]. It provides an expedient access to various useful molecules from readily available olefin substrates. Among the many possible heterodifunctionalization reactions, the addition of a sulfonyl group and oxygen to a double-bond is particularly useful in view of the broad utility of β-oxysulfones (Scheme 1). They are not only useful synthetic intermediates in organic chemistry, but also important substructures in a range of biological active molecules [3]. For example, a β-hydroxysulfone unit is present in both the anti-androgen drug molecule casodex and the antifungal agent SSY726 (Fig. 1) [3b-d].

|
Download:
|
| Scheme 1. Introduction to β-oxysulfonylation of olefins. | |

|
Download:
|
| Fig. 1. Selected bioactive molecules containing a β-oxysulfone unit. | |
Owing to the significance of this subunit, the development of efficient methods to access β-oxysulfones has received tremendous attention in organic synthesis [4, 5]. Among them, olefin oxysulfonylation is one of the most studied processes [4]. In the presence of a suitable oxidant and a source of the sulfonyl group, this process can be achieved with high efficiency under various conditions, such as metal catalysis, metal-free, and even photocatalytic conditions. However, in almost all these processes, a stoichiometric amount of oxidant has to be used due to the oxidation nature of this process. Moreover, in essentially all these examples, a free β-hydroxy group is installed (R = H, Scheme 1). Recently, electrochemical synthesis has emerged as an attractive tool for oxidation reactions that obviates the use of stoichiometric oxidants, which provides a clean and safe alternative to chemical oxidation [6-8]. Herein we describe an electrochemical synthesis of a range of β-oxysulfones in the absence of chemical oxidants via olefin addition of not only hydroxy group, but also alkoxy and carbonyloxy groups (R = H, alkyl, and carbonyl) to the β position of the sulfonyl group.
We began our study with 2-methyl styrene 1a as the model substrate and phenyl sulfinic acid 2a as the sulfonyl source. A mixture of MeCN and water was used as solvent, which also provides the nucleophile for the formation of the desired β-hydroxysulfone product 3a. After considerable efforts in evaluating different electrochemical conditions, we found that the best reaction efficiency was achieved with tetrabutylammonium tetrafluoroborate as electrolyte at a constant current of 10 mA in an undivided cell using platinum plate as the cathode and graphite rod as the anode. With this set of conditions, the desired hydroxysulfone product 3a was formed in 85% NMR yield as the single regio isomer (76% isolated yield, Table 1, entry 1). Variation of the parameters from the standard conditions resulted in inferior efficiency. For example, either a lower current or a higher current led to decreased yield (entries 2 and 3). Using other salts as electrolyte, such as nBu4NPF6, nBu4NClO4, and LiClO4 proved inferior (entries 4–6). Moreover, this process was also found to be sensitive to the loading of phenyl sulfinic acid 2a. Decreasing its loading from 2.5 equiv. to 1 or 2 equiv. led to a lower yield (entries 7 and 8). The ratio of water and MeCN also influenced the reaction outcome significantly. Variation from the optimum ratio (1:4) in either direction also resulted in significantly lower efficiency (entries 9 and 10). Finally, we also found that the graphite anode was also important to the success. Replacing it with a Pt-anode, the product was formed in only 20% yield (entry 11).
|
|
Table 1 Evaluation of the electrochemical conditions.a |

{kind=link}
{kind=link}
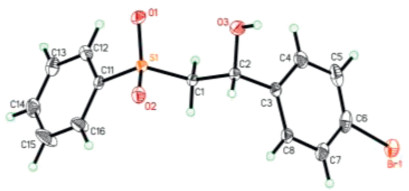
With the standard reaction conditions, we then examined its generality towards different substrates. As shown in Scheme 2, a range of different substituted styrenes successfully participated in this oxysulfonylation process, affording the corresponding vicinal difunctionalization products 3 in moderate to good yields. Monosubstituted and 1, 1-disubstituted olefins were suitable substrates. Unfortunately, internal olefins were not reactive toward the desired products under the standard conditions, presumably due to increased steric hindrance in the C–S bond formation step. Notably, in addition to the formation of β-hydroxysulfones, our protocol is also amenable to the synthesis of β-alkoxysulfones 3f and 3g in the presence of an alcohol (in places of water/MeCN) under otherwise identical conditions. It is worth noting that such an extension was rarely demonstrated in the previous hydroxysulfonylation processes. Finally, the structure of product 3b was confirmed by X-ray crystallography (Fig. 2).

|
Download:
|
| Scheme 2. Reaction scope. Reaction conditions: 1 (0.50 mmol), 2a (1.25 mmol), nBu4NBF4 (0.25 mmol), H2O/MeCN (1:4, 10 mL) or ROH (10 mL) with 5 Å MS (250 mg), undivided cell, graphite rod anode, Pt plate cathode, constant current at 10 mA, room temperature, open air, 4.5 h, isolated yield. | |
{kind=link}

|
Download:
|
| Fig. 2. ORTEP structure of compound 3b (CCDC 1893818). | |
{kind=link}
Inspired by the success in the above three-component intermolecular functionalization process, we were interested in extending it to intramolecular cyclization process (Scheme 3). Thus, a range of styrenes tethered with an internal alcohol or carboxylic acid nucleophile unit were synthesized and subjected to the standard conditions. Gratifyingly, these substrates reacted smoothly with a diverse set of sulfinic acids to form a range of cyclic ethers or lactones bearing an exocyclic sulfonyl functionality [9].

|
Download:
|
| Scheme 3. Intramolecular reaction. Reaction conditions: 1 (0.50 mmol), 2 (1.25 mmol), nBu4NBF4 (0.25 mmol), MeCN (10 mL), undivided cell, graphite rod anode, Pt plate cathode, constant current at 10 mA, room temperature, open air, 4.5 h, isolated yield. | |
{kind=link}
Based on the results, we have proposed a possible reaction mechanism (Scheme 4). Since the redox potential of the styrene substrate is higher than that of the sulfinic acid, we believe that the reaction begins with initial oxidation of the sulfinic acid in the anode to form sulfonyl radical Ⅰ. Subsequent C–S bond formation between this radical and styrene 1 forms benzylic radical Ⅱ, which undergoes an additional oxidation step in the anode to form benzylic cation Ⅲ. In the presence of a nucleophile, such as water, alcohol, and carboxylic acid, new C–O bond formation takes places to form the observed oxysulfone product. In the meanwhile, proton is reduced to hydrogen gas in the cathode to maintain current in the closed circuit.

|
Download:
|
| Scheme 4. Proposed mechanism. | |
{kind=link}
In summary, we have developed an electrochemical vicinal heterodifunctionalization of olefins for the synthesis of β-oxysulfones. With suitable choice of the conditions, including current, electrodes, and electrolyte, this oxidation reaction proceeded efficiently in an undivided cell without the use of a stoichiometric chemical oxidant. In addition to the previously established synthesis of β-hydroxysulfones in the presence of water, minor modification of this protocol by using either external alcohol nucleophiles or internal carboxylic acid nucleophile also led to the synthesis of β-alkoxysulfones, and β-sulfonyl lactones.
AcknowledgmentsFinancial support was provided by Hong Kong RGC (No. 16302318), Shenzhen Science and Technology Innovation Committee (No. JCYJ20170818113708560), and HKUST (No. IEG17SC03). We thank Herman H. Y. Sung for assistance in structure determination by X-ray crystallography.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.023.
| [1] |
(a) H.C. Kolb, M.S. VanNieuwenhze, K.B. Sharpless, Chem. Rev. 94 (1994) 2483-2547; (b) J. Rodriguez, J.P. Dulcère, Synthesis (1993) 1177-1205; (c) B.M. Trost, I. Fleming, M.F. Semmelhack, Comprehensive Organic Synthesis, Pergamon Press, Oxford, 1991; (d) R.I. McDonald, G. Liu, S.S. Stahl, Chem. Rev. 111 (2011) 2981-3019. |
| [2] |
(a) Z. Yuan, H.Y. Wang, X. Mu, et al., J. Am. Chem. Soc. 137 (2015) 2468-2471; (b) H. Chen, A. Kaga, S. Chiba, Org. Biomol. Chem. 14 (2016) 5481-5485; (c) D.B. Bagal, G. Kachkovskyi, M. Knorn, et al., Angew. Chem. Int. Ed. 54 (2015) 6999-7002; (d) Y. An, D. Zheng, J. Wu, Chem. Commun. 50 (2014) 11746-11748; (e) D.F. Lu, C.L. Zhu, J.D. Sears, H. Xu, J. Am. Chem. Soc. 138 (2016) 11360-11367. |
| [3] |
(a) J.E. Baldwin, Sulphones in Organic Synthesis, Vol. 10, Pergamon, 1993; (b) R.A. Fromtling, Drugs Future 14 (1989) 1165-1168; (c) S. Oida, Y. Tajima, T. Konosu, et al., Chem. Pharm. Bull. 48 (2000) 694-707; (d) H. Eto, Y. Kaneko, S. Takeda, et al., Chem. Pharm. Bull. 49 (2001) 173-782. |
| [4] |
(a) Q. Lu, J. Zhang, F. Wei, et al., Angew. Chem. Int. Ed. 52 (2013) 7156-7159; (b) A. Kariya, T. Yamaguchi, T. Nobuta, et al., RSC Adv. 4 (2014) 13191-13194; (c) N. Taniguchi, J. Org. Chem. 80 (2015) 7797-7802; (d) K. Choudhuri, T.K. Achar, P. Mal, Adv. Synth. Catal. 359 (2017) 3566-3576; (e) Y. Wang, W. Jiang, C. Huo, J. Org. Chem. 82 (2017) 10628-10634; (f) T. Taniguchi, A. Idota, H. Ishibashi, Org. Biomol. Chem. 9 (2011) 3151-3153; (g) S.K. Pagire, S. Paria, O. Reiser, Org. Lett. 18 (2016) 2106-2109. |
| [5] |
(a) A.L. Moure, R.G. Arrayás, J.C. Carretero, Chem. Commun. 47 (2011) 6701-6703; (b) A.S. Deeming, C.J. Russell, A.J. Hennessy, M.C. Willis, Org. Lett.16 (2014) 150-153; (c) N. Chumachenko, P. Sampson, Tetrahedron 62 (2006) 4540-4548; (d) S.N.Murthy, B. Madhav, V.P. Reddy, K.R. Rao, Y.V.D. Nageswar, Tetrahedron Lett. 50 (2009) 5009-5011. |
| [6] |
F. Marken, M. Atobe, Modern Electrosynthetic Methods in Organic Chemistry, CRC Press, Taylor & Francis Group, 2019.
|
| [7] |
(a) J.C. Siu, G.S. Sauer, A. Saha, et al., J. Am. Chem. Soc. 140 (2018) 1251112510; (b) L. Zhang, G. Zhang, P. Wang, Y. Li, A. Lei, Org. Lett. 20 (2018) 7396-7399; (c) K.Y. Ye, G. Pombar, N. Fu, et al., J. Am. Chem. Soc. 140 (2018) 2438-3441; (d) M.W. Zheng, X. Yuan, Y.S. Cui, et al., Org. Lett. 20 (2018) 7784-7789; (e) C.Y. Cai, H.C. Xu, Nat. Commun. 9 (2018) 3551; (f) P. Xiong, H. Long, J. Song, et al., J. Am. Chem. Soc. 140 (2018) 16387-16391. |
| [8] |
(a) C.K. Chan, N.C. Lo, P.Y. Chen, M.Y. Chang, Synthesis 49 (2017) 4469-4477; (b) Y.C. Luo, X.J. Pan, G.Q. Yuan, Tetrahedron 71 (2015) 2119-2123. |
| [9] |
Y. Wang, L. Deng, J. Zhou, et al., Adv. Synth. Catal. 360 (2018) 1060-1065. DOI:10.1002/adsc.201701532 |