 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, China

Qingjiang Li was born in Anhui, China in 1985. He received his Bachelor degree in 2007 from Lanzhou University. Then he moved to Peking University and obtained his Ph.D. under the guidance of Prof. Yanxing Jia. After two years as a research assistant in Prof. Henry N.C. Wong's lab at the Chinese University of Hong Kong, he was appointed as an associate professor at Sun Yat-sen University in 2014. His current research interests are the metal-catalyzed C–H functionalization reactions, the development of novel reactions for organic synthesis, and total synthesis of natural products.
Since the early 2000s, transition-metal-catalyzed inert C–H activation reaction [1] has evolved to be an efficient, straightforward and powerful tool for the synthesis of functional organic materials, biologically active molecules, and complex natural products in a highly atom- [2] and step-economic manner [3]. Up to now, the noble second- and third-row transition metals, such as ruthenium, rhodium, palladium, and iridium, have overwhelmingly dominated the developed catalytic C—H functionalization field. However, from the viewpoint of sustainable chemistry, the use of first-row transition metals, such as manganese, iron, cobalt, nickel, and copper, in lieu of noble metals in C–H activation catalysis is highly desirable not only owing to their cost-efficiency and low toxicity but also their potentially unique reactivity. In this regard, unsaturated π bond containing coupling partners such as alkenes and alkynes have been widely studied in 3d transition-metal-catalyzed C–H activation transformations.
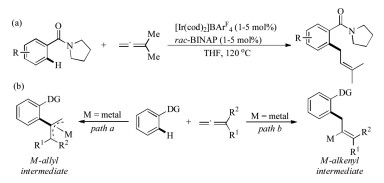
Allene moiety is widespread in a great number of bioactive compounds, functional materials, and naturally occurring products [4]. Owing to the two orthogonal C—C double bonds, allenes have exhibited high reactivity and chemical diversity in modern synthetic transformations [5]. Pioneered by Krische in 2009 (Scheme 1a) [6], and later advanced by the groups of Ma [7], Ackermann [8], Cheng [9], Glorius [10], Cramer [11], Rao [12], and others [13], allene was found to be an interesting coupling partner in transition-metal-catalyzed C–H functionalization reactions. Different from alkenes and alkynes, the versatile reactivities and ample reaction types of allenes in C–H activation chemistry are mainly governed by the formation of either metal-allyl (Scheme 1b, path a) or metal-alkenyl intermediates (Scheme 1b, path b) through the selective insertion of an organometallic species with allene. Since the steric and electronic nature of the substituents on allene could affect the insertion pattern, the selectivity (including regio-, chemo-, and stereoselectivity) is very rich and hard to control in allene participated C–H functionalization reactions.

|
Download:
|
| Scheme 1. Allenes in transition-metal-catalyzed C–H activation. | |
{kind=link}
Since the first report of allene in C–H functionalization via cobalt catalysis disclosed by Cheng and coworkers in 2016 [14], considerable achievements have been made towards the use of first-row transition metals with allenes during the past three years. Whilst recent review about the flexible reactivity of allenes in transition-metal-catalyzed C–H functionalization reactions has been published [15], this review mainly summarized the progress of the first-row transition-metal-catalyzed C–H activation reaction with allenes until the end of 2018. Hence, this review is presented and grouped basically according to the key metal complexes that play an essential role in allene participated 3d transition-metalcatalyzed C–H activation reactions.
2. Manganese-catalyzed C–H functionalization with allenesManganese is the third most abundant transition metal after iron and titanium. Compared to traditional expensive metals such us palladium, platinum, rhodium, ruthenium and iridium, manganese has the characteristics of low toxicity and low cost. It is assessed as a particularly attractive alternative to replace the typically used transition metal catalysts [1f, 1i, 16].
In 2017, we reported the first Mn catalyzed sp2 C–H bond activation of ketimines 1 with allenes 2 (Scheme 2) [17]. In this study, one-pot synthesis of polycyclic products via Mn-catalyzed C—H allylation and Ag-catalyzed Povarov cyclization was accomplished. The reaction featured high bond-forming efficiency, broad substrate scope, good functional group tolerance, high regio- and stereoselectivity, and 100% atom economy. A plausible mechanism was proposed to rationalize the catalytic cycle and stereochemistry. Initially, an imine-assisted C–H activation took place to produce intermediate B. Thereafter, allene coordination and migratory insertion of the terminal double bond gave the intermediate D, then protonation of D generated allylation product E. The selective formation of Z type alkene might rise from the preferential migratory insertion from the less shielded π face of allene. Subsequently, the polycyclic products 3 could be obtained through Ag-catalyzed Povarov cyclization of E.

|
Download:
|
| Scheme 2. Mn/Ag relay catalyzed cyclization of ketimines with allenes [17]. | |
{kind=link}
Later, under the similar catalytic reaction conditions, Mn(Ⅰ)- catalyzed direct C–H coupling with allenes for the efficient assembly of prenylated arenes was described by our group (Scheme 3) [18]. Thus, with the aid of pyridine or pyrimidine directing group, a variety of prenylated heteroarenes 5, such as indole, pyrrole, thiophene, isoquinolin-1(2H)-one, pyridin-2(1H)- one, and 1H-benzo[d]imidazole were obtained in good to excellent yields. Disappointedly, tri- or tetra-substituted allenes were completely unsuccessful for this reaction.

|
Download:
|
| Scheme 3. Mn(Ⅰ)-catalyzed C–H prenylation of various heteroarenes [18]. | |
{kind=link}
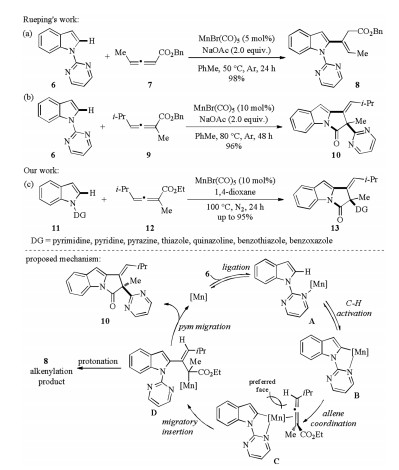
The regio- and stereoselective alkenylation of indoles with 1, 3- disubstituted allenoates under manganese catalysis was developed by Rueping and co-workers in 2017 (Scheme 4a) [19]. Thus, indole 6 reacted with disubstituted allenoate 7 in the presence of MnBr(CO)5 catalyst and NaOAc additive to give alkenylation product 8 in 98% yield. Interestingly, when trisubstituted allenoates were subjected to the reaction conditions, a directing group (DG) migratory annulation product 10 was formed in high efficiency (Scheme 4b) [19]. In the meantime, the directing group migratory annulation reaction was also established by our group using MnBr(CO)5 catalyst in 1, 4-dioxane medium without NaOAc additive (Scheme 4c) [20]. Notably, a series of heterocycles such as pyrimidine, pyridine, pyrazine, thiazole, quinazoline, benzothiazole, and benzoxazole were also applicable to migrate when used as the directing group. Mechanically, the reaction began with DGassisted C–H activation to generate intermediate B followed by regio- and stereoselective migratory insertion to give intermediate D. On one hand, the protonation of D resulted in the formation of alkenylation product 8. On the other hand, the strong nucleophilicity of C-Mn bond could lead to the N-to-C 1, 4-migration of the DG and followed an intramolecular displacement to furnish the final cyclized product 10.

|
Download:
|
| Scheme 4. Mn(Ⅰ)-catalyzed C–H alkenylation and cyclization of indoles with allenes [19, 20]. | |
{kind=link}
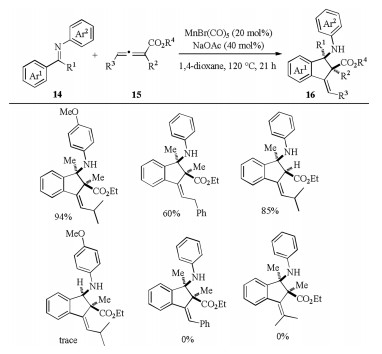
Taking advantage of the strong nucleophilicity [21] of C-Mn bond, Ding and co-workers recently reported C–H alkenylation/annulation reaction of aromatic ketimines 14 with poly-substituted ester-activated allenes 15 under manganese catalysis (Scheme 5) [22]. Of note, this catalytic reaction afforded 2, 3- dihydro-1H-indene products 16 bearing two vicinal all-substituted carbon stereocenters with high diastereoselectivity and E/Z selectivity. The 1, 3-disubstituted allenyl esters were also suitable for the Mn(Ⅰ) catalyzed C–H annulation, however, the employment of C3 phenyl-substituted- or tetrasubstituted allenoate and aldimine would shut down the reaction.

|
Download:
|
| Scheme 5. Mn(Ⅰ)-catalyzed C–H alkenylation/annulation of ketimines with allenes [22]. | |
{kind=link}
Very recently, Glorius et al. disclosed an elegant manganese(Ⅰ)/BPh3-cocatalyzed highly regioselective C–H propargylation of arenes with bromoallenes (Scheme 6) [23]. Lewis acid BPh3 was found to be very important for this transformation. It could not only improve the electrophilicity of bromoallene, but also enhance the regioselectivity of insertion of an organomanganese species with allene. The substrate scope showed good functional group tolerance, thus, secondary, tertiary, and even quaternary carbon centers could be introduced at the propargylic position through this protocol. In addition to internal alkynes, terminal alkynes could also be obtained by this method starting from corresponding bromoallenes. Moreover, the central chirality at the propargylic position could be successfully constructed by chirality transfer from axial chirality in the bromoallene.

|
Download:
|
| Scheme 6. Mn(Ⅰ)/BPh3-cocatalyzed C–H propargylation using bromoallenes [23]. | |
{kind=link}
3. Iron-catalyzed C–H functionalization with allenes
Iron is the least toxic and most earth-abundant transition metal, which also plays a significant role in C–H functionalizations [24]. Recently, Ackermann and co-workers reported the first Fecatalyzed allene annulation through C–H/N—H/C–O/C–H functionalization sequence (Scheme 7) [25]. With the removable dimethylmethylene triazole (TAM) group-assisted Fe-catalyzed C– H activation, the reaction of benzamide 20 with allene 21 gave the exo-methylene dihydroisoquinoline 22 in excellent yield (Scheme 7a). Otherwise, by the use of methylene-tethered triazole (TAH) group, the corresponding isoquinolone 24 was yielded through further isomerization of exo-methylene-3, 4-dihydroisoquinoline. The measured intermolecular KIE value of 1.2 indicated that C–H cleavage was not the rate-limiting step. The authors proposed a possible mechanism that started with iron-catalyzed C–H activation followed by coordination and migratory insertion of the allene to alkenyl ironacycle B. Thereafter, oxidation-induced reductive elimination gave iron allyl species C, which underwent a unique intramolecular 1, 4-iron migration to deliver allylicbenzylic iron intermediate D. Finally, the protonation of the resulting C-Fe bond with the amide motif of the starting material produced exo-methylene dihydroisoquinoline 22 and regenerated active ironacycle species A. Both experimental and computational studies supported the crucial 1, 4-iron migration process involved in this mechanism.

|
Download:
|
| Scheme 7. Iron-catalyzed C–H/N—H/C–O/C–H allene annulation [25]. | |
{kind=link}
4. Cobalt-catalyzed C–H functionalization with allenes
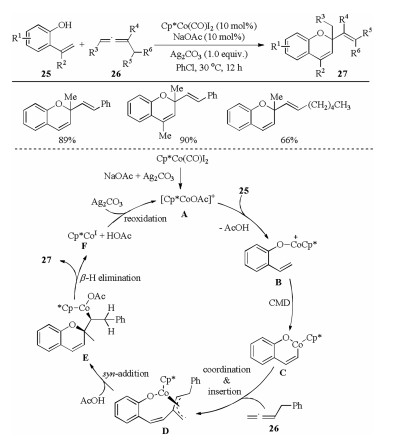
In recent years, the early transition metal cobalt has been emerged as a cheap, environmentally friendly and effective catalyst for a great deal of C–H functionalization reactions [26]. In this context, Cheng et al. reported the first example that allenes were employed as the coupling partners in the cobalt-catalyzed C–H activation reactions in 2016 (Scheme 8) [14]. A Cp*CoⅢ-catalyzed -OH assisted vinylic C–H activation of 2-vinyl phenols 25 with allenes 26 via an oxidative [5 + 1] cyclization to give various 2H-chromenes 27 bearing a valuable exocyclic double bond, in which the allene acted as a one-carbon coupling partner. The use of OAc- containing additives was crucial for this transformation. The approach proceeded in a regio- and stereoselective manner under mild reaction conditions. Mechanism investigations indicated that an intramolecular regio- and stereoselective nuclephilic attack of the O-Co bond to the center carbon of the π-allylic group in D followed by a β-H elimination was involved in the catalytic cycle.

|
Download:
|
| Scheme 8. Cp*CoⅢ-catalyzed [5 + 1] annulation of 2-vinyl phenols with allenes [14]. | |
{kind=link}
The introduction of an alkenyl moiety into molecules is of longstanding interest in organic synthesis. In this respect, Ackermann and co-workers developed a cationic cobalt complex catalyzed C–H alkenylation of arenes with allenes (Scheme 9) [27]. The reaction of arenes 28 with 1, 1-disubstituted allenes 2 in the presence of 5 mol% Cp*Co(CO)I2 and 10 mol% AgSbF6 in 1, 2- dichloroethane gave alkenylated arenes 29 with high chemo- and regioselectivity. Variously substituted arenes and indole derivatives with a wide range of functional groups, including fluoro, chloro, bromo, and ester substituents, smoothly underwent the C–H alkenylation. A bulky alkyl group was essential for the alkenylation reaction, whereas the allylation product was obtained when a di-n-alkyl-substituted allene was used. Disappointedly, mono-, tri-, and 1, 3-disubstituted allenes were not suitable for this transformation.

|
Download:
|
| Scheme 9. Cobalt-catalyzed alkenylation of arenes with allenes [27]. | |
{kind=link}
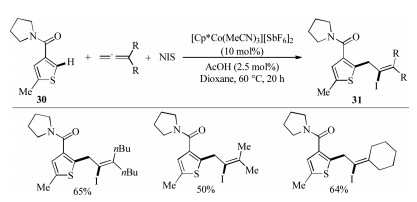
Multicomponent reaction (MCR) provides a sequential manner to give highly selective products that retain majority of the atoms of the starting materials in a single reaction with high efficiency [28]. In 2017, Ellman et al. demonstrated a cationic cobalt species catalyzed three-component C–H coupling of thiophenes with 1, 1- disubstituted allenes and NIS for the synthesis of tetrasubstituted alkenyl iodides with high regioselectivity (Scheme 10) [29]. Notably, alkenyl halides are valuable synthetic handles for further derivatization in metal-catalyzed cross-coupling reactions.

|
Download:
|
| Scheme 10. Cobalt-catalyzed three-component halo-arylation of allenes [29]. | |
{kind=link}
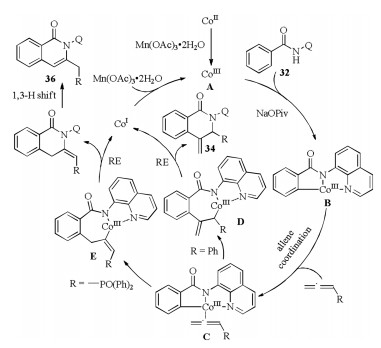
N-Containing heterocyclic skeletons are widespread in naturally occurring products and biologically active molecules and play an important role in medicinal chemistry. Thus, the construction of such compounds starting from simple raw materials by means of C–H activation process is attractive for organic chemists. In 2016, Volla et al. reported a 8-aminoquinoline (AQ) assisted regioselective intermolecular [4 + 2] cyclization of arylamides or alkenylamides 32 with allenes under cobalt catalysis (Scheme 11a) [30]. The regioselectivity might rise from the electronic and steric properties of allenes. When phenylallene 33 was used, the dihydroisoquinolin-1(2H)-one 34 was obtained via reductive elimination of Co-π-allyl intermediate D (Scheme 12). While using the electron deficient allenyl phosphonate 35, isoquinolinone product 36 was formed via reductive elimination of Coalkenyl complex E (Scheme 12) with good efficiency. Later, Rao et al. [12b] and Cheng et al. [31] independently disclosed the similar annulation of arylamides 32 with electron deficient 37 or alkyl substituted allene 40, leading to the formation of dihydroisoquinolin-1(2H)-one 38 or isoquinolinone 41 (Schemes 11b and c), respectively. Notably in Rao's work, the basicity of additive could change the formation of exo- or endo-cyclic isoquinolinones via 1, 3-H shift.

|
Download:
|
| Scheme 11. Co-catalyzed oxidative [4 + 2] annulation of amides with allenes [30, 12b, 31]. | |
{kind=link}

|
Download:
|
| Scheme 12. Mechanism of Co-catalyzed regioselective [4 + 2] annulation [30]. | |
{kind=link}
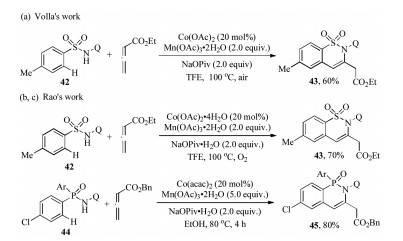
The reaction system also can be extanded to sulfonamide and phophinamide substrates. In 2017, Volla et al. [32] and Rao et al. [12a] independently reported the rapid construction of biologically relevent sultams through cobalt-catalyzed C–H/N—H functionalization of sulfonamides with allenes (Schemes 13a and b). The reaction had a wide range of substrate scope and was tolerant of a variety of functional groups. In addition, a one-pot protocal and gram-scale synthesis were performed to demonstrate the synthetic utility of such annulation. After that, Rao's group further disclosed a facile synthesis of biological phosphaisoquinolin-1-one scaffold 45 via the similar Co-catalyzed [4 + 2] annulation reaction of phophinamides 44 and allenes with good yields and high regioselectivity (Scheme 13c) [12c].

|
Download:
|
| Scheme 13. Co-catalyzed regioselective synthesis of sultams and phosphaisoquinolin-1-ones [32, 12a, 12c]. | |
{kind=link}
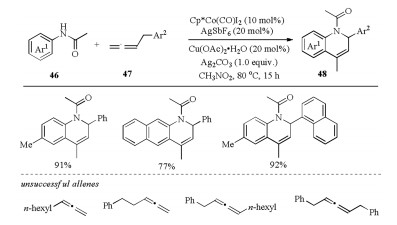
In addition to the commonly encountered formal [4 + 2] and [5 + 1] annulation reactions toward the construction of sixmembered heterocycles, formal [3 + 3] annulation provides an alternative strategy. In 2018, Cheng and coworkers reported the synthesis of 1, 2-dihydroquinolines (1, 2-DHQ) 48 by Cp*Co(Ⅲ)- catalyzed oxidative [3 + 3] annulation of anilides 46 with benzylallenes 47, in which allene acted as a three-carbon synthon in C–H activation process (Scheme 14) [33]. In this transformation, both acetate and AgSbF6 were proven to be important additives for the annulation. Unfortunately, the substrate scope was restricted to terminal benzyl-type allenes, alkyl or internal allenes were not compatible with this method. Of note, the 1, 2-DHQ products 48 could be easily converted to the corresponding quinoline and 1, 2, 3, 4-tetrahydroquinoline.

|
Download:
|
| Scheme 14. CoⅢ-catalyzed formal [3 + 3] annulation of anilides with benzylallenes [33]. | |
{kind=link}
Mechanistic studies indicated that the C–H activation step was reversible and might be involved in the rate limiting step. The proposed mechanism for the Cp*Co(Ⅲ)-catalyzed oxidative [3 + 3] annulation reaction was outlined in Scheme 15. After ortho C–H metalation of anilide 46 with active catalyst A, the successive allene insertion, β-H elimination and reductive elimination gave diene D and CoⅠ. Then, CoⅢ-mediated intramolecular 1, 4-addition of N—Co bond to the diene group provided product 48 and regenerated active CoⅢ species.

|
Download:
|
| Scheme 15. Plausible mechanism for Co-catalyzed formal [3 + 3] annulation [33]. | |
{kind=link}
Very recently, Cheng and co-workers reported a cobalt(Ⅲ)- catalyzed [4 + 1] annulation of amides with allenes for the synthesis of isoindolones and 1, 5-dihydro-pyrrol-2-ones (Scheme 16) [34]. The approach tolerated a wide range of substrates and gave the desired products in good to excellent yields with good regioselectivity. The use of catalytic amount of acetate ion was necessary for this transformation, as it captured the proton in the C—H activation step. However, the use of simple aliphatic and 1, 1-disubstituted allenes did not give the desired annulation products. The mechanistic investigations indicated that the β-H elimination gave the diene intermediate (like D in Scheme 15), which underwent Co-H insertion and reductive elimination to deliver the desired [4 + 1] annulation product.

|
Download:
|
| Scheme 16. Co-catalyzed [4 + 1] annulation of amides with allenes [34]. | |
{kind=link}
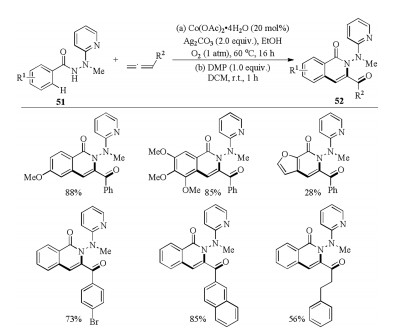
In 2018, Zhai and coworkers disclosed the synthesis of versatile 3-acylquinolines 52 by means of cobalt-catalyzed trifunctionalization of allenes in the presence of molecular oxygen (Scheme 17) [35]. The C–H bond trifunctionalization reaction was assisted by a bidentate 2-(1′-methylhydrazinyl)pyridine (MHP) directing group and proceeded under mild reaction conditions with high bondforming efficiency and good tolerance of a variety of functional groups. A number of commonly encountered functional groups, regardless of the electronic nature, at different positions of phenyl ring of benzoic hydrazides or benzylallenes were well tolerated, giving the corresponding products in generally good to excellent yields. Mechanistic studies strongly suggested that the generated ketone oxygen atom was originated from the dioxygen rather than from moisture or reaction medium EtOH.

|
Download:
|
| Scheme 17. Co-catalyzed trifunctionalization of allenes in the presence of O2 [35]. | |
{kind=link}
In recent years, electrochemistry has a notable application in organic synthesis. In this context, Ackermann et al. reported the first cobalt-catalyzed pyridyl N-oxide directed electrocatalytic C–H/N—H functionalization of (hetero)arenes and alkenes with allenes (Scheme 18) [36]. The reaction featured excellent chemoselectivity, site selectivity, good regioselectivity, and mild reaction conditions. Interestingly, the exo-methylene isoquinolones were obtained by the use of internal allenes. Moreover, mechanistic studies suggested that the C–H bond cleavage was not involved in the rate-determining step and was assisted by carboxylate.

|
Download:
|
| Scheme 18. Electrooxidative allene annulations via Co-catalyzed C–H activation [36]. | |
{kind=link}
5. Nickel-catalyzed C–H functionalization with allenes
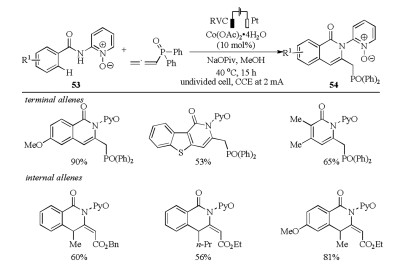
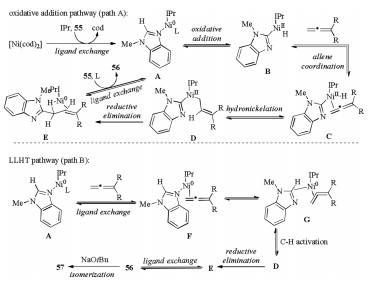
In 2017, Ackermann's group reported a nickel(0)-catalyzed C–H allylation and alkenylation of imidazole and purine derivatives with allenes without directing group (Scheme 19) [37]. The reaction of heterocycles 55 with 1, 1-disubstituted allenes in the presence of 10 mol% Ni(cod)2 and 10 mol% IPr (1, 3-bis(2, 6- diisopropylphenyl)imidazol-2-ylidene) in toluene afforded the allylation products 56, whereas the alkenylation products 57 were delivered by the addition of stoichiometric amounts of NaOtBu. The NHC ligand also played an important role in this transformation. The reaction tolerated a broad range of heterocycles and allenes in a highly regio- and stereoselective manner with good to excellent yields. Importantly, the synthetic potential of this reaction was demonstrated by the powerful late-stage C–H alkenylation of diphosphodiesterase inhibitors. Mechanistic studies supported the first nickel(0)-catalyzed C–H activation with allenes occurred through two possible pathways including oxidative addition and ligand-to-ligand-hydrogen transfer (LLHT) to form key Ni(Ⅱ) species and a KIE value of kH/kD = 1.1 suggested the C–H cleavage was not involved in the rate limiting step (Scheme 20).

|
Download:
|
| Scheme 19. Nickel-catalyzed C–H allylation and alkenylation with allenes [37]. | |
{kind=link}

|
Download:
|
| Scheme 20. Plausible mechanism for Ni-catalyzed hydroarylations of allenes. | |
{kind=link}
Initially, the active [NHC-Ni0Ln] complex A was generated through a ligand exchange, which was followed by successive oxidative addition with the C–H bond, coordination and migratory addition of the Ni-C bond to the terminal double bond of the allene, and reductive elimination to give the product-nickel complex E (path A). Further ligand exchange with substrate 55 would deliver the desired allylated product 56 and regenerate the complex A. Alternatively, complex A firstly coordinated with the allene, then the corresponding intermediate G underwent C–H activation through ligand-to-ligand-hydrogen transfer to furnish key intermediate D (path B). Allylated product 56 could isomerize into the alkenylated product 57 in the presence of NaOtBu (Scheme 20).
6. ConclusionIn conclusion, recent advances of allenes in the first-row transition metals catalyzed C–H activation reactions have been summarized. These developments have showcased the fascinating and interesting reactivities of allenes in metal-catalyzed C–H functionalizations. Futher studies in this area will help to gain more insights into additional new reactivities of allenes, particularly for functionalized and tetrasubstituted allenes. In addition to common regio- and chemoselectivity issues, the first-row transition-metal-catalyzed C–H functionalization with allenes in an enantioselective manner should be a challenging topic in this field. Moreover, the exploration of untouched 3d transition metals catalyzed sp3 C–H activation with allenes would largely expand the substrates scope for complex molecules synthesis.
7. AcknowledgmentsWe are grateful for the support of this work by the National Natural Science Foundation of China (No. 21502242) and the State Key Laboratory of Natural and Biomimetic Drugs (No. K20150215).
| [1] |
(a) P. Gandeepan, T. Müller, D. Zell, et al., Chem. Rev. 119 (2019) 2192-2452; (b) Y. Hu, B. Zhou, C. Wang, Acc. Chem. Res. 51 (2018) 816-827; (c) S. Prakash, R. Kuppusamy, C.H. Cheng, ChemCatChem 10 (2018) 683-705; (d) Y. Xu, G. Dong, Chem. Sci. 9 (2018) 1424-1432; (e) J. He, M. Wasa, K.S.L. Chan, Q. Shao, J.Q. Yu, Chem. Rev. 117 (2017) 8754-8786; (f) J.R. Hummel, J.A. Boerth, J.A. Ellman, Chem. Rev. 117 (2017) 9163-9227; (g) Y. Park, Y. Kim, S. Chang, Chem. Rev. 117 (2017) 9247-9301; (h) J. Jiao, K. Murakami, K. Itami, ACS Catal. 6 (2016) 610-633; (i) W. Liu, L. Ackermann, ACS Catal. 6 (2016) 3743-3752; (j) T. Gensch, M.N. Hopkinson, F. Glorius, J. Wencel-Delord, Chem. Soc. Rev. 45 (2016) 2900-2936; (k) F. Wang, S. Yu, X. Li, Chem. Soc. Rev. 45 (2016) 6462-6477; (l) M. Gulías, J.L. Mascareñas, Angew. Chem. Int. Ed. 55 (2016) 11000-11019; (m) B. Ye, N. Cramer, Acc. Chem. Res. 48 (2015) 1308-1318; (n) O. Daugulis, J. Roane, L.D. Tran, Acc. Chem. Res. 48 (2015) 1053-1064; (o) B. Liu, F. Hu, B.F. Shi, ACS Catal. 48 (2015) 1863-1881; (p) S.A. Girard, T. Knauber, C.J. Li, Angew. Chem. Int. Ed. 53 (2014) 74-100; (q) G. Rouquet, N. Chatani, Angew. Chem. Int. Ed. 52 (2013) 11726-11743; (r) X. Shang, Z.Q. Liu, Chem. Soc. Rev. 42 (2013) 3253-3260; (s) B.J. Li, Z.J. Shi, Chem. Soc. Rev. 41 (2012) 5588-5598; (t) L. McMurray, F. O'Hara, M.J. Gaunt, Chem. Soc. Rev. 40 (2011) 1885-1898; (u) C.S. Yeung, V.M. Dong, Chem. Rev. 111 (2011) 1215-1292; (v) T. Newhouse, P.S. Baran, Angew. Chem. Int. Ed. 50 (2011) 3362-3374; (w) T.W. Lyons, M.S. Sanford, Chem. Rev. 110 (2010) 1147-1169. |
| [2] |
B.M. Trost, Science 254 (1991) 1471-1477. DOI:10.1126/science.1962206 |
| [3] |
P.A. Wender, B.L. Miller, Nature 460 (2009) 197-201. DOI:10.1038/460197a |
| [4] |
(a) A. Hoffmann-Röder, N. Krause, Angew. Chem. Int. Ed. 43 (2004) 1196-1216; (b) P. Rivera-Fuentes, F. Diederich, Angew. Chem. Int. Ed. 51 (2012) 2818-2828; (c) S. Yu, S. Ma, Angew. Chem. Int. Ed. 51 (2012) 3074-3112. |
| [5] |
(a) B. Yang, Y. Qiu, J.E. Bäckvall, Acc. Chem. Res. 51 (2018) 1520-1531; (b) J. Ye, S. Ma, Acc. Chem. Res. 47 (2014) 989-1000; (c) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria, A. Simonneau, Chem. Rev. 111 (2011) 1954-1993; (d) S. Ma, Chem. Rev. 105 (2005) 2829-2872; (e) A.S.K. Hashmi, Angew. Chem. Int. Ed. 39 (2000) 3590-3593; (f) N. Krause, A.S. Hashmi, Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004. |
| [6] |
Y.J. Zhang, E. Skucas, M.J. Krische, Org. Lett. 11 (2009) 4248-4250. DOI:10.1021/ol901759t |
| [7] |
(a) Z. Fang, C. Fu, S. Ma, Chem. -Eur. J. 16 (2010) 3910-3913; (b) R. Zeng, C. Fu, S. Ma, J. Am. Chem. Soc. 134 (2012) 9597-9600; (c) R. Zeng, S. Wu, C. Fu, S. Ma, J. Am. Chem. Soc. 135 (2013) 18284-18287. |
| [8] |
S. Nakanowatari, L. Ackermann, Chem. -Eur. J. 21 (2015) 16246-16251. DOI:10.1002/chem.201502785 |
| [9] |
P. Gandeepan, P. Rajamalli, C.H. Cheng, Chem. -Eur. J. 21 (2015) 9198-9203. DOI:10.1002/chem.201501106 |
| [10] |
(a) H. Wang, F. Glorius, Angew. Chem. Int. Ed. 51 (2012) 7318-7322; (b) H. Wang, B. Beiring, D.G. Yu, K.D. Collins, F. Glorius, Angew. Chem. Int. Ed. 52 (2013) 12430-12434. |
| [11] |
(a) D.N. Tran, N. Cramer, Angew. Chem. Int. Ed. 49 (2010) 8181-8184; (b) D.N. Tran, N. Cramer, Angew. Chem. Int. Ed. 52 (2013) 10630-10634; (c) B. Ye, N. Cramer, J. Am. Chem. Soc. 135 (2013) 636-639. |
| [12] |
(a) T. Lan, L. Wang, Y. Rao, Org. Lett. 19 (2017) 972-975; (b) T. Li, C. Zhang, Y. Tan, W. Pan, Y. Rao, Org. Chem. Front. 4 (2017) 204-209; (c) X. Yao, L. Jin, Y. Rao, Asian J. Org. Chem. 6 (2017) 825-830. |
| [13] |
(a) Y. Kuninobu, P. Yu, K. Takai, Org. Lett. 12 (2010) 4274-4276; (b) X.F. Xia, Y.Q. Wang, L.L. Zhang, et al., Chem.-Eur. J. 20 (2014) 5087-5091; (c) T.J. Gong, W. Su, Z.J. Liu, et al., Org. Lett. 16 (2014) 330-333. |
| [14] |
R. Kuppusamy, K. Muralirajan, C.H. Cheng, ACS Catal. 6 (2016) 3909-3913. DOI:10.1021/acscatal.6b00978 |
| [15] |
R. Santhoshkumar, C.H. Cheng, Asian J. Org. Chem. 7 (2018) 1151-1163. DOI:10.1002/ajoc.201800133 |
| [16] |
C. Wang, Synlett 24 (2013) 1606-1613. DOI:10.1055/s-0033-1339299 |
| [17] |
S.Y. Chen, Q. Li, X.G. Liu, et al., ChemSusChem 10 (2017) 2360-2364. DOI:10.1002/cssc.201700452 |
| [18] |
S.Y. Chen, Q. Li, H. Wang, J. Org. Chem. 82 (2017) 11173-11181. DOI:10.1021/acs.joc.7b02220 |
| [19] |
C. Wang, A. Wang, M. Rueping, Angew. Chem. Int. Ed. 56 (2017) 9935-9938. DOI:10.1002/anie.201704682 |
| [20] |
S.Y. Chen, X.L. Han, J.Q. Wu, et al., Angew. Chem. Int. Ed. 56 (2017) 9939-9943. DOI:10.1002/anie.201704952 |
| [21] |
(a) Y. Kuninobu, Y. Nishina, T. Takeuchi, K. Takai, Angew. Chem. Int. Ed. 46 (2007) 6518-6520; (b) B. Zhou, Y. Hu, C. Wang, Angew. Chem. Int. Ed. 54 (2015) 13659-13663; (c) W. Liu, J. Bang, Y. Zhang, L. Ackermann, Angew. Chem. Int. Ed. 54 (2015) 14137-14140; (d) Y.F. Liang, L. Massignan, W. Liu, L. Ackermann, Chem. -Eur. J. 22 (2016) 14856-14859; (e) S. Sueki, Z. Wang, Y. Kuninobu, Org. Lett. 18 (2016) 304-307; (f) B. Zhou, Y. Hu, T. Liu, C. Wang, Nat. Commun. 8 (2017) 1169-1177. |
| [22] |
C. Lei, L. Peng, K. Ding, Adv. Synth. Catal. 360 (2018) 2952-2958. DOI:10.1002/adsc.201800465 |
| [23] |
C. Zhu, J.L. Schwarz, S. Cembellín, S. Greβies, F. Glorius, Angew. Chem. Int. Ed. 57 (2018) 437-441. DOI:10.1002/anie.201710835 |
| [24] |
(a) P. Wang, L. Deng, Chin. J. Chem. 36 (2018) 1222-1240; (b) R. Shang, L. Ilies, E. Nakamura, Chem. Rev. 117 (2017) 9086-9139; (c) F. Jia, Z. Li, Org. Chem. Front. 1 (2014) 194-214. |
| [25] |
J. Mo, T. Müller, J.C.A. Oliveira, L. Ackermann, Angew. Chem. Int. Ed. 57 (2018) 7719-7723. DOI:10.1002/anie.201801324 |
| [26] |
(a) T. Yoshino, S. Matsunaga, Adv. Synth. Catal. 359 (2017) 1245-1262; (b) S. Wang, S.Y. Chen, Chem. Commun. 53 (2017) 3165-3180. |
| [27] |
S. Nakanowatari, R. Mei, M. Feldt, L. Ackermann, ACS Catal. 7 (2017) 2511-2515. DOI:10.1021/acscatal.7b00207 |
| [28] |
J.P. Wan, L. Gan, Y. Liu, Org. Biomol. Chem. 15 (2017) 9031-9043. DOI:10.1039/C7OB02011B |
| [29] |
J.A. Boerth, J.A. Ellman, Angew. Chem. Int. Ed. 56 (2017) 9976-9980. DOI:10.1002/anie.201705817 |
| [30] |
N. Thrimurtulu, A. Dey, D. Maiti, C.M.R. Volla, Angew. Chem. Int. Ed. 55 (2016) 12361-12365. DOI:10.1002/anie.201604956 |
| [31] |
R. Boobalan, R. Kuppusamy, R. Santhoshkumar, P. Gandeepan, C.H. Cheng, ChemCatChem 9 (2017) 273-277. DOI:10.1002/cctc.201601190 |
| [32] |
N. Thrimurtulu, R. Nallagonda, C.M.R. Volla, Chem. Commun. 53 (2017) 1872-1875. DOI:10.1039/C6CC08622E |
| [33] |
R. Kuppusamy, R. Santhoshkumar, R. Boobalan, H.R. Wu, C.C. Cheng, ACS Catal. 8 (2018) 1880-1883. DOI:10.1021/acscatal.7b04087 |
| [34] |
R. Boobalan, R. Santhoshkumar, C.H. Cheng, Adv. Synth. Catal. 361 (2019) 1140-1145. DOI:10.1002/adsc.201801335 |
| [35] |
S. Zhai, S. Qiu, X. Chen, et al., ACS Catal. 8 (2018) 6645-6649. DOI:10.1021/acscatal.8b01720 |
| [36] |
T.H. Meyer, J.C.A. Oliveira, S.C.S. Sau, N.W.J. Ang, L. Ackermann, ACS Catal. 8 (2018) 9140-9147. DOI:10.1021/acscatal.8b03066 |
| [37] |
S. Nakanowatari, T. Müller, J.C.A. Oliveira, L. Ackermann, Angew. Chem. Int. Ed. 56 (2017) 15891-15895. DOI:10.1002/anie.201709087 |