 2019, Vol. 30
2019, Vol. 30 



Zhenhua Jia received his bachelor degree from Tianjin University and obtained his pH.D. from Sun Yat-sen University with Prof. Albert. S. C. Chan, jointed with Prof. Chao-Jun Li at McGill University in Canada. After a postdoctoral stay with Prof. Chae S. Yi at Marquette University, U.S., he worked in Prof. Henry N.C. Wong's lab in Chinese University of Hong Kong as a research assistant. Subsequently, he moved to University of Alberta as a Mitacs research fellow with Prof. Fred. G. West, where he investigated photo-promoted organic transformations toward sustainable chemistry. In 2018, he accepted his current position at Nanjing Tech University as full Professor. His research focuses on the discovery of unconventional chemical reactivity and efficient approaches to organic transformation, as well as Biomass transformation.
In modern organic chemistry, a sustainable transformation plays a key role to minimize the consumption of energy and chemical reagents.Although Ciamician pioneered the photochemistry for over 100 years [1], the photon has been sought as a more effective and sustainable energy input to overcome the energy barrier in the phase of activation, which is also considered as a traceless reagent. Photocatalys is is introduced to describe the chemical process, in which the rate of a reaction is changed or initiated under the radiation of ultraviolet, visible or infrared light in the presence of a substance that absorbs the energy of light and is involved in the chemical transformation of the reaction partners [2]. During the last decade, visible light-induced photo-catalysis in the area of synthetic organic chemistry is developed rapidly. Recently, Noyori recommended perspectives of green chemistry to develop a 'photo-synthetic' catalyst that facilitates a thermally unachievable, energeticallyuphill reaction in future [3]. The majority of the photo-catalysts used nowadays are complexes of transition-metal centered complexes coordinated with elegant ligands [4]. Under the irradiation of visible light, the excited photo-catalysts promote the process of singleelectron-transfer (SET) or energy transfer (ET).
Accompanying with the application of metal-based photocatalysts in organic synthesis, the properties of organometallic complexes are critical to preparing biologically active compounds, such as potential toxicities and separation problems from final products to meet high levels of purity, and emerging concerns have further increased. Alternatively, organic dyes as photo-catalysts are typically less expensive, widely available, and easy to handle and modify. Moreover, organic photo-catalysts may have properties that canout-performmetal-basedphoto-catalysts.Sinceorganicdyesare applied in photo-catalysis some decades ago, the demand for more sustainable methods has also triggered considerable progress in the field of metal-free visible light photo-catalysis. Nicewicz and Kokotos documented well the topics in organic photo-catalysis respectively [5, 6]. In their reviews, they summarized the progress of organic photoredox catalysis, involving conventional organic dyes as photo-sensitizers, such as cyanoarenes, benzophenones, quinones, pyryliums, thiapyryliums, quinoliniums, acridiniums, fluoresceins, rhodamines, phenothiazines, thioxanthones, phenylglyoxylic acids, and other organic photoredox catalysts.
This mini-review will focus on the recent advance of literature regarding photo-promoted transition metal-free organic transformations in the absence of conventional photo-sensitizers. In particular, it will focus on photo-promoted simple ketones-induced organic transformations, photo-promoted hypervalent iodide-mediated organic transformations and the more recent advance of photopromoted organic transformations under catalyst-free conditions.
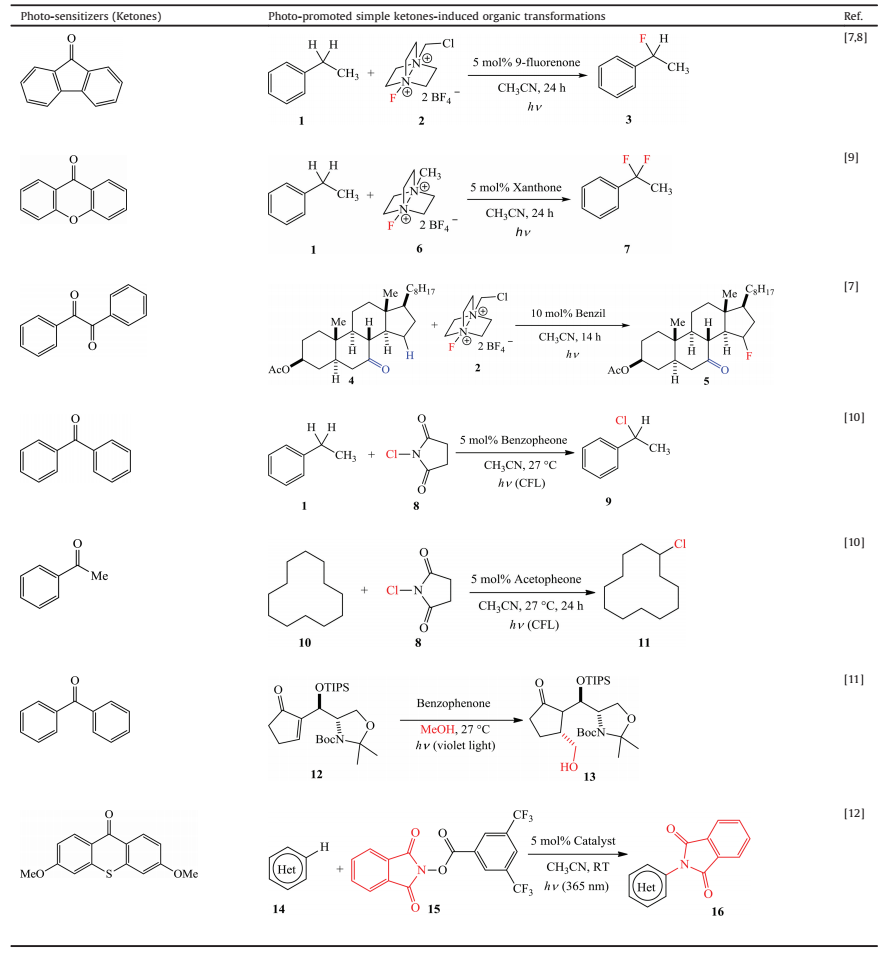
2. Photo-promoted simple ketones-induced organic transformationsSince Chen et al. reported visible light can activate simple diarylketones to induce mono- and difluorination of benzylic C—H bonds selectively in 2013 [7], this strategy was highlighted in organic photo-catalysis many times. Simple ketones as unconventional photo-sensitizers were attracted attention to be treated as initiators to induce organic transformations. Subsequently, Frutos and Kappe et al. improve the efficiency of Chen's methodology via a continuous-flow protocol [8]. Chen's fluorination was also suited for complex, polycyclic molecules. Lectka et al. utilized ketones as directing groups to initiate sp3 C—H fluorination when benzil was activated by cool white light to trigger this transformation [9]. Subsequently, Chen's group achieved kinetic chlorination of C(sp3)-H bonds. For simple substrates, chlorination products can be generated with good yields. However, for more complex substrates the regioselectivity of chlorination is still lower to be further improved in future [10]. More recently, Chen developed a protocol of C—H functionalization mediated by triplet ketone to rapid access to the core skeleton of the [3 + 2] type dimeric pyrroleimidazole alkaloids with moderate yield and regioselectivity [11]. Ooi et al. reported a photoexcited aryl ketone could catalyze C—H imidation of arenes and heteroarenes combined with an easy obtained imidating reagent. They further applied this protocol to the direct C—H acyloxylation of arenes. Based on experimental and theoretical approaches, they disclosed the probable mechanism that the thioxanthone derived catalyst acts as an excited-state single-electron reductant and establishes an oxidative quenching cycle (Table 1) [12].
|
|
Table 1 Chen's initial strategy and relevant organic transformations. |
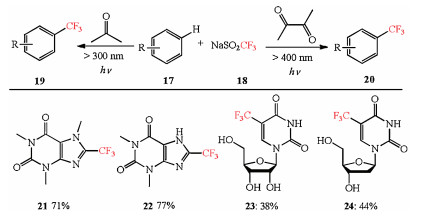
In 2016, Li et al. demonstrated simple aliphatic ketones, such as acetone and diacetyl, can be used as radical initiators under ultraviolet or visible light irradiation to generate CF3 radicals efficiently from sodium triflate. The scope of this strategy is broad and the direct trifluoromethylations are achieved, when xanthine alkaloids (21, 22) and antiviral drug molecules (23, 24) as substrates were exposed under the irradiation of different lights, which depends on the ketones they used as solvent or cosolvent (Scheme 1) [13].

|
Download:
|
| Scheme 1. Li's aromatic trifluoromethylation. | |
{kind=link}
3. Photo-promoted hypervalent iodide-mediated organic transformations
In 2014, Morokuma et al. succeeded to generate the acyl radicals from branched aldehydes by using hypervalent iodine(Ⅲ) catalysts under mild visible-light photolysis, which were subsequently trapped with some electrophilic olefins. They disclosed a rare example, in which the acyl radicals were initiated by a catalytic amount of hypervalent iodine(Ⅲ) reagent under irradiation of visible light (Scheme 2) [14].

|
Download:
|
| Scheme 2. Morokuma's C—H activation and acylation reaction. | |
{kind=link}
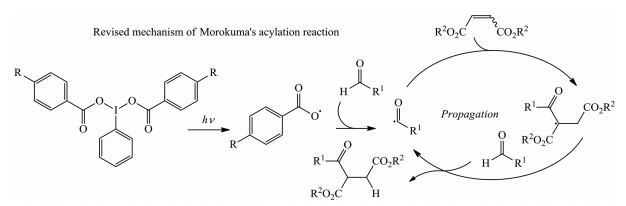
Subsequently, they revised their previous proposed mechanism in details via experimental observations and computational calculations. The revised mechanism demonstrated that the catalytic reagent of hypervalent iodine is the initiator of the radical reaction. The formation of acyl radical is rate-determining by kinetic isotope effect (KIE) experiment and the resulting radical acts as the chain carrier for the process of propagation (Scheme 3) [15].

|
Download:
|
| Scheme 3. Revised mechanism of Morokuma's acylation reaction. | |
{kind=link}
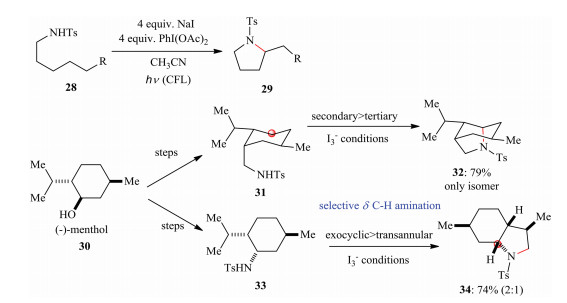
Nagib et al. reported a strategy of selective distal Cδ-H amination to access a broad range of biologically relevant pyrrolidines in 2016. This novel intramolecular cyclization is mediated by a triiodide (I3-). They observed unique selectivities when they started from the menthol as starting material to prepare the intermediates (28, 29) that were further transformed under triiodide (I3-) conditions (Scheme 4) [16].

|
Download:
|
| Scheme 4. Nagib's distal C—H amination. | |
{kind=link}
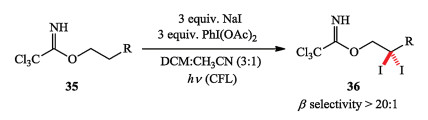
Based on the previous strategy, the same group first achieved a radical relay protocol for mono- and di-halogenation (including iodination, bromination, and chlorination). In particular, the versatile and geminal di-halogenation was uniquely facilitated by an iterative radical -based 1, 5-HAT (hydrogen atom transfer) to abstract sequential sp3 C–H bonds (Scheme 5) [17].

|
Download:
|
| Scheme 5. Nagib's di-halogenation reaction. | |
{kind=link}
4. Photo-promoted organic transformations under catalyst-free conditions
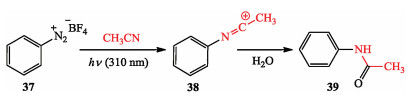
The photo can also promote organic transformations under catalyst-free conditions in the absence of conventional photosensitizers, including photoredox catalysts. In 2003, Albini et al. reported the photo-heterolysis of para-substituted diazonium tetrafluoroborates in MeCN leads to the singlet phenyl cations which add the solvent to yield the corresponding acetanilides avoiding reduction of diazonium cation (Scheme 6) [18].

|
Download:
|
| Scheme 6. Albini's photo-heterolysis of para-substituted diazonium tetrafluoroborates. | |
{kind=link}
They extended the scope of this photo-heterolysis strategy to generate the triplet phenyl cations from para-substituted diazonium tetrafluoroborates to phenyl sulfonates and phosphates substituted with electron-donating groups. Compared with metal catalysis, reaction conditions are more mild and insensitive to oxygen and moisture, also not involving the use of bases or other aggressive reagents (Scheme 7) [19].

|
Download:
|
| Scheme 7. Albini's photo-generation of the triplet phenyl cations. | |
{kind=link}
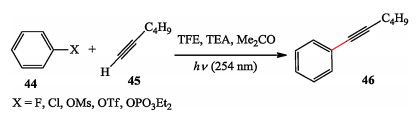
To evaluate the further application of photo-generation of the triplet aryl cations, they developed a photo-promoted Sonogashira cross-coupling reaction of electron-rich aryl chlorides and aryl esters with alkynes under transition metal-free conditions (Scheme 8) [20].

|
Download:
|
| Scheme 8. Albini's photo-promoted Sonogashira cross-coupling reaction. | |
{kind=link}
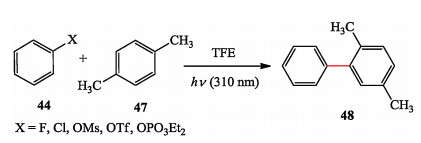
They also developed a protocol of photochemical synthesis of biaryl compounds. In traditional palladium catalyzed crosscoupling reactions, aryl chlorides, and even aryl fluorides and phosphates are generally less reactive, but these precursors are well accommodated under photochemical conditions (Scheme 9) [21].

|
Download:
|
| Scheme 9. Albini's photo-arylation reaction. | |
{kind=link}
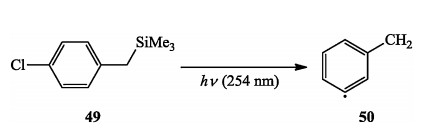
Recently, Albini et al.discovered an alternative method to achieve α, n-didehydrotoluenes by photoactivation of chlorobenzyl-trimethylsilanes, which were trapped with overcoming the limitation of conventional enyne-allenes cyclization (Scheme 10) [22].

|
Download:
|
| Scheme 10. Albini's photo-generation of α, n-didehydrotoluenes. | |
{kind=link}
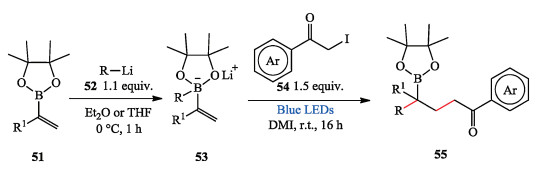
In 2017, Aggarwal et al. reported a novel photo-induced 1, 2- metalate rearrangement, which was triggered via oxidation of an α-boronate radical. This radical species can be generated from a halide and a vinyl boronate induced by visible light, although the use of a photoredox catalyst can enhance yields significantly in some cases. This method covered vast substrate scope providing a novel, efficient and atom-economic three-component coupling reaction. The scope of the radical precursor includes α-iodo ketones, esters, nitriles, primary amides, α-fluorinated haloacetates, and perfluoroalkyl iodides. Furthermore, many of the products are simply not accessible through traditional 1, 2- metalate rearrangements (Scheme 11) [23].

|
Download:
|
| Scheme 11. Aggarwal's photo-induced 1, 2-metalate rearrangements. | |
{kind=link}
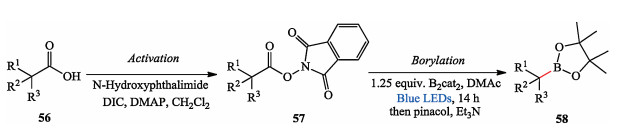
Afterward, they achieved photo-induced decarboxylative borylation of carboxylic acids. They found that this transformation can be effected by irradiating the N-hydroxyphthalimide ester derivatives of the carboxylic acids with visible light at room temperature in the presence of the diboron reagent bis(catecholate)diboron (B2cat2), demonstrating broad utility of carboxylic acids and great functional group tolerance. After a simple workup, the pinacol boronic esters were isolated (Scheme 12) [24].

|
Download:
|
| Scheme 12. Aggarwal's photo-induced decarboxylative borylation. | |
{kind=link}
More recently, Studer et al. reported photo-induced metal-free borylation of alkyl and aryl iodides with bis(catecholate)diboron (B2cat2) as the boron source under mild conditions. From radical clock experiments and density functional theory calculations, they disclosed the mechanism and rate constants for C-radical borylation with B2cat2 (Scheme 13) [25].

|
Download:
|
| Scheme 13. Studer's photo-induced metal-free borylation. | |
{kind=link}
The borylation reaction was initiated by light-mediated C—I bond homolysis of iodide to generate the alkyl/aryl radical (A), which added to B2cat2 to give the radical (B). Then, the radical (B) was trapped by DMF in an exothermic process to give (C), which readily homolyzed to give boronated product along with the DMFcomplexed boryl radical (D/E). The chain was closed by an I-atom transfer reaction of the iodide via (F) to eventually give (G) and radical (A) (Scheme 14).

|
Download:
|
| Scheme 14. Studer's proposed mechanism. | |
{kind=link}
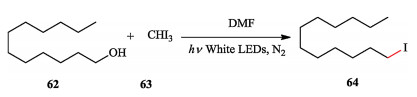
Antonietti and Zhao discovered the alcohol iodination reaction was promoted under the irradiation of visible light. The reaction initiated with the homolysis of the C—I bond of iodoform induced by visible light, producing active radicals, which were trapped by DMF, following nucleophilic substitution to form Vilsmeier-type reagent. The alcohol was iodinated by Vilsmeier-type reagent to generate the product (Scheme 15) [26].

|
Download:
|
| Scheme 15. Antonietti and Zhao's photo-induced alcohol iodination. | |
{kind=link}
Li et al. described a simple and efficient strategy to convert phenol derivatives into aryl radicals via transition-metal-free manner. They accessed aryl radicals successfully using aryl triflates as precursors under photo-irradiation, which were trapped by bis (pinacolato)diboron (B2pin2) and sodium iodide (NaI) to generate two types of important organic molecules, aryl boronates and aryl iodides (Scheme 16) [27].

|
Download:
|
| Scheme 16. Li's photo-induced borylation and iodination. | |
{kind=link}
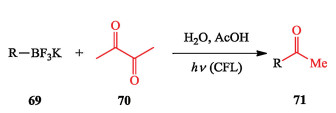
Morerecently, Li et al. established a distinct strategy togenerate both aryl and alkyl radicals with organotrifluoroborates as the precursors via the bimolecular homolytic substitution (SH2) process. This strategy is enabled by using water as the solvent and visible light as the energy input and diacetyl as the promoterin the absence of any metal catalysts or redox reagents, thereby eliminating the metal waste (Scheme 17) [28].

|
Download:
|
| Scheme 17. Li's photo-induced bimolecular homolytic substitution (SH2) process. | |
{kind=link}
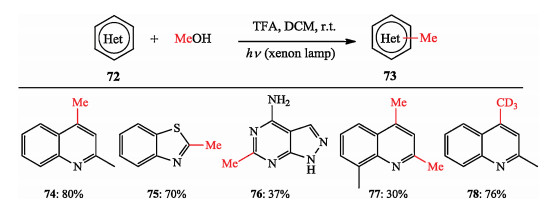
Toward more sustainable organic transformations, Li et al. developed a metal-free methylation protocol promoted by light. They found cheap, readily available, and abundant MeOH as both the solvent and the methylating reagent. Kinds of heteroarenes, bearing various functional groups could be methylated, bismethylated and tri-deuteromethylated in moderate to good yields (Scheme 18) [29].

|
Download:
|
| Scheme 18. Li's photo-induced metal-free methylation. | |
{kind=link}
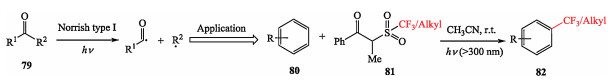
In 2017, Li et al. presented a redox-neutral and catalyst-free protocol to generate trifluoromethyl and alkyl radicals in the context of C—H functionalization of arenes. This protocol, via the Norrish type Ⅰ concept to produce alkyl radicals, accommodates various functional groups and delivers the product in good yields. This method identified a series of compounds as the trifluoromethylation and alkylation reagents assisted by light (Scheme 19) [30].

|
Download:
|
| Scheme 19. Li's photo-induced trifluoromethylation and alkylation via Norrish type Ⅰ approach. | |
{kind=link}
5. Conclusion and perspective
In terms of sustainable chemistry, the efficient capture and utilization of solarenergy is a keychallenge facing chemical society. The exploration of unconventional organic transformations, which can directly access potentially valuable compounds from readily available feedstocks without the need of extensive additives or expensive reagents could have a positive impact on resources, energy, and the environment.Recently, renewed interest has focused on organic transformations via photo-catalysis. Remarkable progress has been made in this field, the examples presented in this mini-review illustrate the recent advance of photo-promoted transition metal-free organic transformations in the absence of conventional photo-sensitizers. During the processes of photoactivation, the metal complexes are excluded to achieve the specific transformations, which are environmentally friendly and economic approaches to avoid causing serious environmental concerns and costissues.But without well-decorated photo-sensitizers to absorb the light, ground-state molecules were difficult to afford their excited states, as a result, the activation is requisite for the short wavelength of light.
Toward sustainability in the future, we envision a great development of photo-promoted unconventional organic transformations, including the development of enantioselective protocols to natural product synthesis and medicinal chemistry. Along with the considerable prospects of renewable energies for organic synthesis and catalysis, major advances are expected in this rapidly evolving photo-promoted organic transformations under the transition metal free conditions.
AcknowledgmentsWe gratefully acknowledge the financial support from the Startup Grant of Nanjing Tech University (No. 38037037), the SICAM Fellowship from Jiangsu National Synergetic Innovation Center for Advanced Materials.
| [1] |
G. Giamician, Science 36 (1912) 385-394. DOI:10.1126/science.36.926.385 |
| [2] |
M. Nic, J. Jirat, B. Kosata, IUPAC Gold Book: Photocatalysis, 3rd. ed., (2006) Oxford.
|
| [3] |
R. Noyori, Tetrahedron (2010) 1028. |
| [4] |
C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322-5363. DOI:10.1021/cr300503r |
| [5] |
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075-10166. DOI:10.1021/acs.chemrev.6b00057 |
| [6] |
I.K. Sideri, E. Voutyritsa, C.G. Kokotos, Org. Biomol. Chem. 16 (2018) 4596-4614. DOI:10.1039/C8OB00725J |
| [7] |
J.B. Xia, C. Zhu, C. Chen, J. Am. Chem. Soc. 135 (2013) 17494-17500. DOI:10.1021/ja410815u |
| [8] |
D. Cantillo, O. De Frutos, J.A. Rinco'n, C. Mateos, C.O. Kappe, J. Org. Chem. 79 (2014) 8486-8490. DOI:10.1021/jo5016757 |
| [9] |
D.D. Bume, C.R. Pitts, F. Ghorbani, et al., Chem. Sci. 8 (2017) 6918-6923. DOI:10.1039/C7SC02703F |
| [10] |
L. Han, J.B. Xia, L. You, C. Chen, Tetrahedron 73 (2017) 3696-3701. DOI:10.1016/j.tet.2017.05.008 |
| [11] |
L. You, C. Chen, Tetrahedron 74 (2018) 769-772. DOI:10.1016/j.tet.2017.12.027 |
| [12] |
C.B. Tripathi, T. Ohtani, M.T. Corbett, T. Ooi, Chem. Sci. 8 (2017) 5622-5627. DOI:10.1039/C7SC01700F |
| [13] |
L. Li, X. Mu, W. Liu, et al., J. Am. Chem. Soc. 138 (2016) 5809-5812. DOI:10.1021/jacs.6b02782 |
| [14] |
S.A. Moteki, A. Usui, S. Selvakumar, T. Zhang, K. Maruoka, Angew. Chem. Int. Ed. 53 (2014) 11060-11064. DOI:10.1002/anie.201406513 |
| [15] |
J. Jiang, R. Ramozzi, S. Moteki, et al., J. Org. Chem. 80 (2015) 9264-9271. DOI:10.1021/acs.joc.5b01695 |
| [16] |
E.A. Wappes, S.C. Fosu, T.C. Chopko, D.A. Nagib, Angew. Chem. Int. Ed. 55 (2016) 9974-9978. DOI:10.1002/anie.201604704 |
| [17] |
E.A. Wappes, A. Vanitcha, D.A. Nagib, Chem. Sci. 9 (2018) 4500-4504. DOI:10.1039/C8SC01214H |
| [18] |
S. Milanesi, M. Fagnoni, A. Albini, Chem. Commun. 9 (2003) 216-217. |
| [19] |
M. De Carolis, S. Protti, M. Fagnoni, A. Albini, Angew. Chem. Int. Ed. 44 (2005) 1232-1236. DOI:10.1002/anie.200461444 |
| [20] |
S. Protti, M. Fagnoni, A. Albini, Angew. Chem. Int. Ed. 44 (2005) 5675-5678. DOI:10.1002/anie.200501541 |
| [21] |
V. Dichiarante, M. Fagnoni, A. Albini, Angew. Chem. Int. Ed. 46 (2007) 6495-6498. DOI:10.1002/anie.200701462 |
| [22] |
S. Protti, D. Ravelli, B. Mannucci, A. Albini, M. Fagnoni, Angew. Chem. Int. Ed. 51 (2012) 8577-8580. DOI:10.1002/anie.201202794 |
| [23] |
M. Silvi, C. Sandford, V.K. Aggarwal, J. Am. Chem. Soc. 139 (2017) 5736-5739. DOI:10.1021/jacs.7b02569 |
| [24] |
A. Fawcett, A. Fawcett, J. Pradeilles, et al., Science 357 (2017) 283-286. DOI:10.1126/science.aan3679 |
| [25] |
Y. Cheng, C. Mück-Lichtenfeld, A. Studer, Angew. Chem. Int. Ed. 57 (2018) 16832-16836. DOI:10.1002/anie.201810782 |
| [26] |
Y. Zhao, M. Antonietti, ChemPhotoChem 2 (2018) 720-724. DOI:10.1002/cptc.201800084 |
| [27] |
W. Liu, X. Yang, Y. Gao, C.J. Li, J. Am. Chem. Soc. 139 (2017) 8621-8627. DOI:10.1021/jacs.7b03538 |
| [28] |
W. Liu, P. Liu, L. Lv, C.J. Li, Angew. Chem. Int. Ed. 57 (2018) 13499-13503. DOI:10.1002/anie.201807181 |
| [29] |
W. Liu, X. Yang, Z.Z. Zhou, C.J. Li, Chem. 2 (2017) 688-702. DOI:10.1016/j.chempr.2017.03.009 |
| [30] |
P. Liu, W. Liu, C.J. Li, J. Am. Chem. Soc. 139 (2017) 14315-14321. DOI:10.1021/jacs.7b08685 |