 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China
Transition metal catalyzed cross coupling between a nucleophile and an electrophile [1], such as Suzuki-Miyaura coupling, Negishi coupling, Kumada coupling, Stille coupling, Sonogashira coupling, powerful protocols for constructing various chemical bonds [2], had been widely used in synthesis of pharmaceuticals [3], nature products [4] and organic materials [5]. The mechanism of the traditional coupling reaction using halide as an electrophile usually involved oxidative addition of transition metal catalyst onto an electrophilic halogen-carbon bond, followed by transmetalation with a nucleophilic organometallic partner and then reductive elimination [6]. Various transition metal complexes such as Pd, Zn, Co, Rh, Ru, Mo, Ni, Cu and Fe were developed as catalysts [3a, 7], allowing for the applications of these coupling reactions in industrial large scale synthesis with mild reaction conditions, excellent functional group compatibility and high yields. Recently, more and more efforts have been focused on expansion the scope of nucleophile and electrophile [8]. Compared to the scope of nucleophile largely extended with various reagents, from very reactive organometallic compounds [2e, 9] to stable boronic acids [1a, 1e, 10], enlarging the scope of electrophile was full of challenges and restricted to halogen derivatives for a long time [1e, 11]. Since halides were expensive and their preparations were often laborious, recently many chemists made great efforts to develop other electrophilic coupling partners instead of halides [12]. Organosulfur compounds such as sulfides, sulfoxides, thioethers, aryl sulfones, sulfoximines, sulfonium salts, thiol esters, sulfamates, sulfonates and arylsulfonyl hydrazides had been used as electrophiles for cross coupling reactions to construct C—C bond [13]. Oxygenous organic compounds like alkenyl acetates, benzylic carbamates, aryl pivalates were also suitable electrophiles for constructing C—C bond [14]. And the cross coupling using primary benzamides, ammonium tetrafluoroborate salts, benzenediazonium, o-benzene disulfonimide and N-boc-amides as electrophiles were also well developed for building C—C, C–O, C–N and C—B bonds [15]. Widely available arenes [16] were also efficient for constructing C—C bond. These above electrophilic alternatives of halides were efficient for constructing various chemical bonds, but there were still some drawbacks remained such as poor functional group compatibility and difficult preparation of starting materials. Development of more efficient electrophilic coupling partners was still in high need.
Nitroarenes, a class of highly versatile, readily available and low toxic building blocks in organic synthesis, would be conveniently and directly obtained by Friedel-Crafts nitration of the parent arenes with good selectivity and high yields in mono-functionalization [17]. In sharp contrast to nitration, the halogenation of arenes often afforded a mixture of mono-, di- and polyhalogenated arenes. Additionally, many commonly used aryl halides were prepared from the corresponding nitroarenes [18]. In nucleophilic substitution reactions of nitroarenes especially those bearing other electron-withdrawing groups, nitro (NO2) group could serve as a leaving group, and this indicated that under certain conditions oxidative addition of metal catalyst onto C–NO2 bond could occur.
For above mentioned advantages of nitroarenes, the direct use of nitroarenes as electrophilic coupling partners instead of aryl halides in transition metal catalyzed cross coupling got more and more attention. In this review, transition metal catalyzed cross coupling of nitroarenes was presented. And nitroarenes were efficient electrophiles for building C–O, C–S, C–N and C—C bonds in the presence of transition metal catalyst.
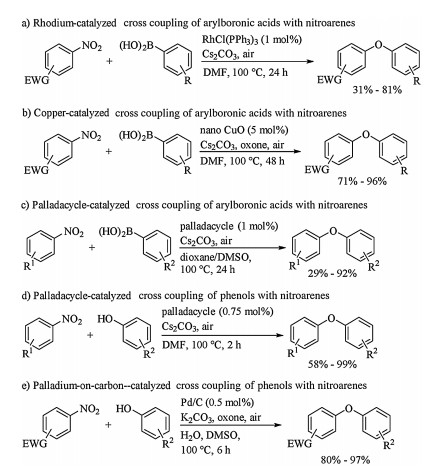
2. Cross coupling of nitroarenes to form C–O bondEarly in 2011, Chen and Wu reported rhodium catalyzed coupling of nitroarenes with aryboronic acids (Scheme 1a) [19]. The reaction proceeded smoothly to afford the desired products in poor to good yields when 1 mol% amount of tris(triphenylphosphine)rhodium chloride (RhCl(PPh3)3) was used as catalyst and 1.25 equiv. amount of cesium carbonate (Cs2CO3) was applied as base. Later, the same researching group developed an improved coupling reaction of nitroarenes for C–O bond construction using nano copper oxide (CuO) as catalyst (Scheme 1b) [20]. In 2013, formation of C–O bond by 2, 2′-bipyridine-palladacycle catalyzed cross coupling of nitroarenes with arylboronic acids was developed by Yu and Chang (Scheme 1c) [21]. Additionally, coupling of nitroarenes with phenols instead of arylboronic acids was also efficient for building C–O bond when palladacycle (Scheme 1d) [22] or Pd/C (Scheme 1e) [23] was used as catalyst and Cs2CO3 was applied as base. In palladacycle catalyzed cross coupling of nitroarenes with phenols (Scheme 1d), 0.75mol% catalyst was enough to obtain reasonable yield, and increasing the loading of catalyst did not improve the yield significantly. These aforementioned protocols were powerful to form C–O bond, but only nitroarenes bearing electron-withdrawing groups gave the desired coupling products in good yields, and the substrate scope need to be expanded. Moreover, a general catalytic system of these reactions for various nitroarenes substrate, especially electronrich ones, remained undeveloped because of difficult oxidative addition of low valent metal onto Ar-NO2 bond.

|
Download:
|
| Scheme 1. Formation of C–O bond by cross coupling of nitroarenes. | |
{kind=link}
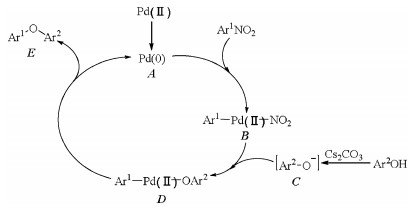
The plausible mechanism for palladacycle catalyzed cross coupling of phenols with nitroarenes was shown in Scheme 2 [22]. The first step of this cross coupling was the generation of active catalyst A, Pd(0). Second, oxidative insertion of catalyst A to Ar1-NO2 bond formed palladium species B. Intermediate D was afforded through reaction of B and C which was obtained by reaction of phenol with Cs2CO3. Finally, D proceeded reductive elimination to give the desired coupling product and regenerate the palladium catalyst.

|
Download:
|
| Scheme 2. Plausible mechanism for palladacycle catalyzed cross coupling of phenols with nitroarenes. | |
{kind=link}
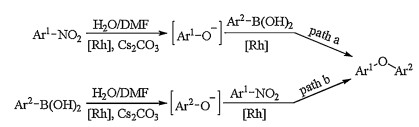
Two plausible pathways were deduced for the rhodium catalyzed cross coupling of arylboronic acids with nitroarene (Scheme 3) [19]. In pathway A, in the presence of RhCl(PPh3)3 and Cs2CO3, water molecule attacked nitroarene to generate the phenoxide intermediate which proceeded cross coupling with arylboronic acid to produce the desired coupling product. Alternatively in pathway B, phenoxide intermediate was generated by oxidative hydroxylation of arylboronic acid in the presence of Cs2CO3. This phenoxide intermediate was then converted to product by nucleophile displacement of NO2 group. And the oxygen atom of product was derived from ambient water, which was confirmed by the reaction of 1-(4-nitrophenyl)ethanone with phenylboronic acid in the presence of 18OH2 in dry DMF.

|
Download:
|
| Scheme 3. Plausible mechanism for rhodium catalyzed cross coupling of arylboronic acids with nitroarenes. | |
{kind=link}
3. Cross coupling of nitroarenes to form C–S bond
In 2013, Shinde reported the first example for copper catalyzed coupling of nitroarenes with thiols (Scheme 4a) [24], in which 20 mol% amount of copper iodide (CuI) in combination with 2 equiv. potassium carbonate (K2CO3) without a ligand in DMF solvent was used as an inexpensive catalytic system. And a series of non-symmetrical aryl thioethers were obtained successfully in excellent yields under mild conditions. The first example for palladium catalyzed coupling of nitroarenes with sulfinates was developed by Yu and Wu in 2014 (Scheme 4b) [25]. Aryl and heterocyclic sulfones were produced in moderate to excellent yields with wide functional group compatibility when 0.75 mol% cyclopalladated ferrocenylimine and 1 equiv. K2CO3 was used as catalyst. In this reaction, 0.75 mol% catalyst was enough for achieving good yield, and increasing the loading of catalyst shew no significant change in yield. Rostami and Ghaderi developed copper catalyzed coupling of nitroarenes with alkyl/benzyl halides, triphenyltin chloride or arylboronic acids for constructing C–S bond in the presence of sulfur (S) sources (Scheme 4c–e) [26]. When 30 mol% Cu(OAc)2 was employed as catalyst and 3 equiv. Cs2CO3 was used as base and 1.5 equiv. sodium thiosulfate pentahydrate (Na2S2O3-5H2O) was used as S source, nitroarenes reacted readily with alkyl or benzyl halide to afford products unsymmetrical sulfides in good to excellent yields. Treatment of nitroarenes with Cu(OAc)2, K2CO3, S8/potassium fluoride (KF), and triphenyltin chloride gave the corresponding unsymmetrical sulfides in excellent yields. In the presence of CuI, S8 and sodium hydroxide (NaOH), nitroarenes reacted with arylboronic acid to form unsymmetrical sulfides.

|
Download:
|
| Scheme 4. Formation of C–S bond by cross coupling of nitroarenes. | |
{kind=link}
The mechanism for palladacycle catalyzed cross coupling of sulfinates with nitroarenes also involved oxidative addition and reductive elimination as shown in Scheme 5 [25]. Intermediate G was formed by oxidative insertion of pre-activated catalyst F to Ar1-NO2 bond, then transmetalation of G with sodium sulfinates produced H. Reductive elimination of H gave the desired coupling product and regenerated the palladium catalyst.

|
Download:
|
| Scheme 5. Plausible mechanism for palladacycle catalyzed cross coupling of sulfinates with nitroarenes. | |
{kind=link}
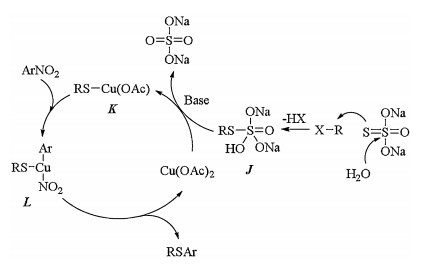
The proposed mechanism for Cu(OAc)2 catalyzed cross coupling reactions of nitroarenes with alkyl/benzyl halides using Na2S2O3-5H2O as S source was shown in Scheme 6 [26]. Treatment of Na2S2O3 with alkyl halide and water generated alkyl thiosulfate J. Oxidative addition of K which was generated by the reaction of J and Cu(OAc)2 to nitroarene produced intermediate L, which transformed to the desired coupling product and released catalyst copper acetate via reductive elimination.

|
Download:
|
| Scheme 6. Plausible mechanism for Cu(OAc)2 catalyzed cross coupling of nitroarenes with alkyl/benzyl halides. | |
{kind=link}
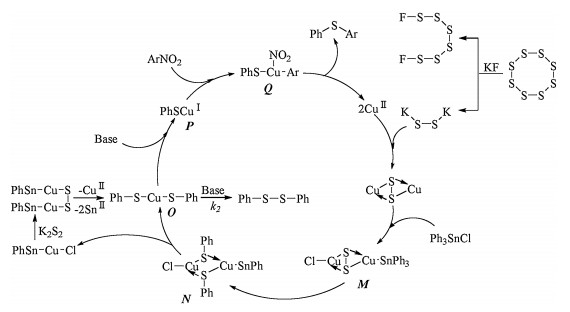
A possible mechanism shown in Scheme 7 was deduced for Cu(OAc)2 catalyzed cross coupling of nitroarene with triphenyltin chloride using S8/KF as S source [26]. Potassium disulfide (K2S2) which was produced from the reaction of S8 with KF reacted with Cu(OAc)2 to give copper sulfide (CuS), which was transformed to intermediate M by oxidative addition to triphenyltin chloride. Intermediate N generated from M via phenyl group immigration was dissociated to intermediate O which could be also generated from N by reacting with K2S2 followed by eliminating copper and tin. O could be transformed to intermediate P or 1, 2-diphenyldisulfane in the presence of base, and the base played a key role at this stage. In the presence of K2CO3 and nitroarene, formation of intermediate P is much quicker than production of 1, 2-diphenyldisulfane (k1≫k2). When triethylamine (Et3N) was used instead of K2CO3, diphenyldisulfide was yielded exclusively (k2≫k1) even when nitroarene existed. Oxidative addition of P to nitroarene formed Q, which underwent reductive elimination to afford the desired phenylarylsulfide and regenerate Cu(Ⅱ) catalyst.

|
Download:
|
| Scheme 7. Plausible mechanism for Cu(OAc)2 catalyzed cross coupling of nitroarenes with triphenyltin chloride. | |
{kind=link}
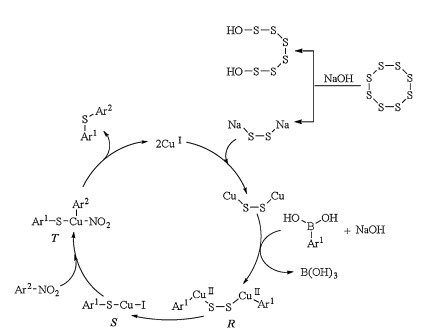
The possible mechanism presented in Scheme 8 was for the coupling of arylboronic acids with nitroarenes in the presence of S8 [26]. Similarly to the mechanism for coupling of nitroarenes with triphenyltin chloride, intermediate R was formed from oxidative addition of arylboronic acid with copper disulfide (Cu2S2) which was generated from the reaction of CuI with sodium disulfide (Na2S2). Key intermediate T was obtained by reaction of nitroarene with S which was produced from R via C–N cleavage. After reductive elimination, T was transformed to the desired diarylsulfide and finished the catalytic cycle.

|
Download:
|
| Scheme 8. Plausible mechanism for Cu(OAc)2 catalyzed cross coupling of arylboronic acids with nitroarenes. | |
{kind=link}
4. Cross coupling of nitroarenes to form C—C bond
Cross coupling reactions for C—C bond formation such as Suzuki-Miyaura coupling, Heck coupling, could offer a straightforward and highly versatile way to access a wide range of functionalized biaryls, olefins and so on [1c]. However formation C—C bond by cross coupling of nitroarenes was rarely developed and reported due to the inherent difficulty associated with oxidative addition of low valent transition metal catalyst to ArNO2 bond and the lack of alternative C–NO2 bond activation mechanism [18].
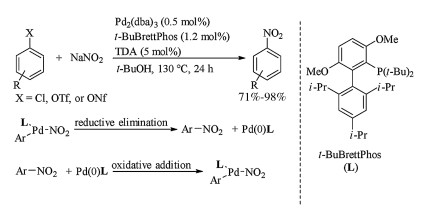
In 2009, Fors and Buchwald reported conversion reaction of aryl chlorides and triflates to nitroarenes using palladium catalyst combining with a bulky monodentate phosphine ligand, t-BuBrettPhos (Scheme 9) [27]. Subjection of aryl chlorides, triflates, or nonaflates and sodium nitrite to Pd2(dba)3/t-Bu-BrettPhos catalytic system produced nitroarenes in good to excellent yields. This nitration protocol was very convenient for preparation nitroarenes with high yields, wide functional group compatibility and mild reaction conditions. The ligand was important in that conversion, and both di-tert-butylphosphino group and methoxy substituent ortho to the phosphine ligand were critical to the reactivity of this catalytic system. Some other ligands like t-Bu-XPhos and XPhos did not give the desired nitroarenes. It was believed that the use of bulky biarylphosphine ligand like t-Bu-BrettPhos (L) ccelerated the reductive elimination of (L)Pd(Ⅱ)Ar(NO2) which formed nitroarenes. Since oxidative addition of Pd(0) to nitroarene was the microscopically reverse process of (L)Pd(Ⅱ)Ar(NO2) reductive elimination, the bulky biarylphosphine ligand could also facilitated the oxidative addition of Pd(0)(L) to nitroarene. These results suggested the utility of bulky biarylphosphine ligands in transition metal catalyzed cross coupling of nitroarenes for constructing C—C bond.

|
Download:
|
| Scheme 9. Conversion reaction of aryl chlorides and triflates to nitroarenes. | |
{kind=link}
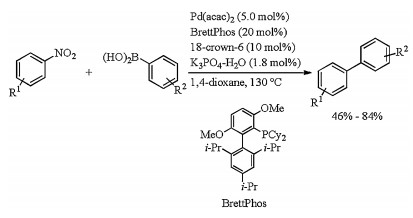
In 2017, Sakaki and Nakao developed palladium catalyzed Suzuki-Miyaura coupling of nitroarenes (Scheme 10) [28]. Biaryls were obtained in moderate to good yields by stirring the mixture of nitroarenes, arylboronic acids, Pd(acac)2, BrettPhos, 18-crown-6, and K3PO4-nH2O in 1, 4-dioxane at 130 ℃ for 24 h. Phosphine ligand was found to be the key for successful implementation of this cross coupling. BrettPhos which was a bulky biarylphosphine ligand with ortho-methoxy substituent gave the best yield, whereas Buchwald's ligands like SPhos, RuPhos, and CPhos were found to be ineffective. Ligands PCy3, Pt-Bu3, and IPr, which were effective for Suzuki-Miyaura coupling of aryl halides and arene sulfonates, did not provide any desired coupling products.

|
Download:
|
| Scheme 10. Formation of C—C bond by cross coupling of nitroarenes. | |
{kind=link}
The following reaction of (cod)Pd(CH2SiMe3)2 with nitrobenzene in the presence of BrettPhos was carried out to gain insight into the reaction mechanism for the above coupling reaction (Scheme 11) [28]. Treatment of (cod)Pd(CH2SiMe3)2 with BrettPhos and nitrobenzene in THF at 60 ℃ produced BrettPhosPd(Ph)(NO2) (U) via oxidative addition of a Pd(0) center onto Ph-NO2 bond. This addition product U was characterized by 1H NMR and 31P NMR spectroscopy, IR spectroscopy, and elemental analysis. The molecular structure of U which was determined by single-crystal X-ray diffraction was similar to that derived from the oxidative addition of aryl halides with the Pd center coordinated not only by the phosphorus atom but also by the triisopropylphenyl ring of BrettPhos. Since U reacted with bromoarenes to give arylpalladium bromides, the oxidative addition could be reversible. The arene-Pd(0) specie, η2-arene complex, whose arene ligand coordinated to Pd through the π-bonds was also a resting state in the catalytic cycle because η2-arene complex V was formed when 1-nitronaphthalene was used instead of nitrobenzene. And then, U underwent transmetalation with (4-methoxyphenyl)boronic acid at 25 ℃ in the presence of K3PO4-nH2O and a subsequent reductive elimination to afford 4-methoxy-1, 1′-biphenyl in 51% yield (GC yield). Thus, the mechanism for Suzuki-Miyaura coupling of nitroarenes should involve oxidative addition, transmetalation and reductive elimination, which was also supported by the DFT calculations.

|
Download:
|
| Scheme 11. Reaction of (cod)Pd(CH2SiMe3)2 with nitrobenzene. | |
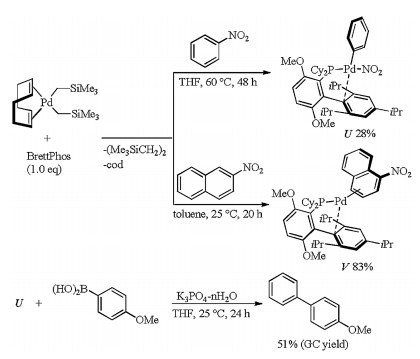{kind=link}
The catalytic cycle shown in Scheme 12, taking coupling of nitrobenzene with phenylboronic acid as an example, was for palladium catalyzed Suzuki-Miyaura coupling of nitroarenes [28]. X generated from the reaction of Pd(acac)2, BrettPhos and nitrobenzene underwent oxidative addition of Pd(0) center to Ph-NO2 bond and afforded intermediate U. In the presence of K3PO4, transmetalation of U with phenylboronic acid gave intermediate W. Reductive elimination of W should afford the desired biaryl compounds and regenerated X. The rate determining step could be the oxidative addition of Pd(0) center to Ar-NO2 bond (In this example is Ph-NO2) in η2-arene complex (In this example is X).

|
Download:
|
| Scheme 12. Plausible mechanism for palladium catalyzed Suzuki-Miyaura coupling of nitrobenzene with phenylboronic acid. | |
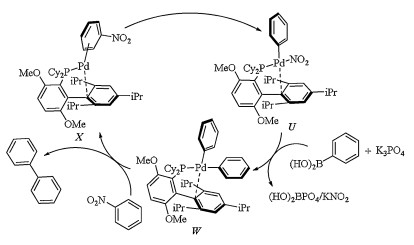{kind=link}
5. Cross coupling of nitroarenes to form C–N bond
In 2017, Nakao reported the first Buchwald-Hartwig amination of nitroarenes (Scheme 13) [29]. The cross coupling of nitroarenes with diarylamines, arylamines, and alkylamines afforded the desired coupling products successfully in moderate to good yields when 5 mol% amount of Pd(acac)2 was employed as catalyst, 15 mol% BrettPhos was used as ligand, and 3 equiv. K3PO4 was played as base. The ligand played a key role in this reaction, ligand BrettPhos gave the best yield, and XPhos also furnished the coupling product in moderate yield. While some phosphine ligands conventionally used in Buchwald-Hartwig amination such as DPPF, BINAP, and P(t-Bu)3 could not give the desired products. The bases also effected the yields dramatically, and strong bases like t-BuOK and t-BuONa commonly used in amination also did not produce the desired products. The above coupling was very powerful for constructing C–N bond, but the yields need to improve.

|
Download:
|
| Scheme 13. Formation of C–N bond by cross coupling of nitroarenes. | |
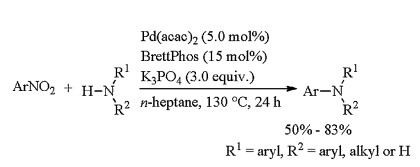{kind=link}
A plausible reaction mechanism for Buchwald-Hartwig amination of nitroarenes was described in Scheme 14 [29]. The key intermediate Y was formed by oxidative addition of Pd(0) to the C–NO2 bond in (L)Pd(0)Ph(NO2) which was generated from the reaction of Pd(acac)2, BrettPhos and Ph-NO2. Subsequently, in the presence of K3PO4 an amine nucleophile reacted with Y to afford arylpalladium amide Z, which could undergo reductive elimination to generate the desired coupling product and arene ligand exchange to regenerate palladium(0) complex.

|
Download:
|
| Scheme 14. Plausible mechanism for Buchwald-Hartwig amination of nitroarenes. | |
{kind=link}
In summary, transition metal catalyzed cross coupling of nitroarenes had attracted more and more attention because nitroarenes were easier available and lower toxic than halides and some other electrophilic coupling partners. Cross coupling of nitroarenes with arylboronic acids or phenols was very powerful for constructing C–O bond. In the presence of S sources, copper catalyzed cross coupling of nitroarenes with alkyl/benzyl halides, triphenyltin chloride or arylboronic acid was efficient to construct C–S bond. Formation of C—C and C–N bonds was also achieved through palladium catalyzed Suzuki-Miyaura coupling of nitroarenes with arylboronic acid and Buchwald-Hartwig amination of nitroarenes with amines.
6. Limitations and perspectivesAlthough these above coupling reactions were useful and convenient for building various chemical bonds, some limitations existed such as expensive transition metal catalysts like rubidium and palladium complexes were often used to realize reasonable yields, poor functional group tolerance and so on. Thus, establishment of cheap and general catalytic system should be focused in future research. And application of nitroarenes electrophilic coupling partners in other cross coupling reactions was still in high need.
AcknowledgmentsThis paper is dedicated to Professor Henry N.C. Wong for his significant contribution to the development of organic chemistry in China. We gratefully acknowledge the National Natural Science Foundation of China (Nos. 21802040, 21877034), the Natural Science Fund Youth Project of Hunan Province (No. 2018JJ3145), the General project of Hunan Education Department (No. 17C0629), and the Open Foundation of Key Laboratory of Theoretical Organic Chemistry and Functional Molecule of Ministry of Education, Hunan University of Science and Technology (No. E21843) for financial support.
| [1] |
(a) N. Miyaura, A. Suzuki, Chem. Rev. 95 (1995) 2457-2483; (b) B. Zhang, R. Breslow, J. Am. Chem. Soc. 119 (1997) 1676-1681; (c) Ei. Negishi, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 1998; (d) A. Minato, K. Suzuki, K. Tamao, M. Kumada, J. Chem. Soc. Chem. Commun. 8 (1984) 511-513; (e) A.F. Littke, G.C. Fu, Angew. Chem. Int. Ed. 41 (2002) 4176-4211; (f) A.R. Chinchilla, C. Nájera, Chem. Rev. 107 (2007) 874-922; (g) L.F. Peng, S.W. Zhang, B.H. Wang, et al., Chin. J. Org. Chem. 38 (2018) 519-525. |
| [2] |
(a) X. Li, J.W. Han, H.N.C. Wong, Asian J. Org. Chem. 5 (2016) 74-81; (b) K.C. Nicolaou, P.G. Bulger, D. Sarlah, Angew. Chem. Int. Ed. 44 (2005) 4442-4489; (c) L.F. Peng, B.H. Wang, M. Wang, et al., J. Chem. Res. 42 (2018) 235-238; (d) L.F. Peng, J.Y. Lei, L. Wu, et al., J. Chem. Res. 42 (2018) 271-273; (e) L.F. Peng, C. Peng, M. Wang, et al., Chin. J. Org. Chem. 38 (2018) 3048-3055; (f) R. Jana, T.P. Pathak, M.S. Sigman, Chem. Rev. 111 (2011) 1417-1492. |
| [3] |
(a) J. Magano, J.R. Dunetz, Chem. Rev. 111 (2011) 2177-2250; (b) T.E. Barder, S.D. Walker, J.R. Martinelli, S.L. Buchwald, J. Am. Chem. Soc. 127 (2005) 4685-4696; (c) S. Abbas, L. Ferris, A.K. Norton, Org. Process Res. Dev. 12 (2008) 202-212; (d) S. Yu, A. Haight, B. Kotecki, et al., J. Org. Chem. 74 (2009) 9539-9542; (e) J.Q. Wan, Y. Xia, Y. Liu, et al., J. Med. Chem. 52 (2009) 1144-1155. |
| [4] |
(a) M.S. Karatholuvhu, A. Sinclair, A.F. Newton, et al., J. Am. Chem. Soc. 128 (2006) 12656-12657; (b) Q. Su, L.A. Dakin, J.S. Panek, J. Org. Chem. 72 (2007) 2-24; (c) V.V. Vintonyak, B. Kunze, F. Sasse, M.E. Maier, Chem.-Eur. J.14 (2008) 11132-11140; (d) G. Dyker, D. Hildebrandt, J. Org. Chem. 70 (2005) 6093-6096. |
| [5] |
(a) C.K. Hau, S.S.Y. Chui, W. Lu, Chem. Sci. 2 (2011) 1068-1075; (b) C.L. Deng, X.D. Xiong, D.T.W. Chik, Chem.-Asian J. 10 (2015) 2342-234; (c) Y. Yamaguchi, T. Ochi, T. Wakamiya, Y. Matsubara, Z. Yoshida, Org. Lett. 8 (2006) 717-720; (d) J.N. Wilson, M. Josowicz, Y. Wang, U.H.F. Bunz, Chem. Commun. (2003) 2982-2983; (e) H. Sugiura, Y. Nigorikawa, Y. Saiki, K. Nakamura, M. Yamaguchi, J. Am. Chem. Soc. 126 (2004) 14858-14864; (f) T.L. Wu, H.H. Chou, P.Y. Huang, C.H. Cheng, R.S. Liu, J. Org. Chem. 79 (2014) 267-274; (g) J. Kim, A.R. Han, J. Hong, et al., Chem. Mater. 26 (2014) 4933-4942; (h) P. Deria, C.D. Von Bargen, J.H. Olivier, et al., J. Am. Chem. Soc. 135 (2013) 16220-16234; (i) Z.Y. Yao, M. Zhang, H. Wu, et al., J. Am. Chem. Soc. 137 (2015) 3799-3802. |
| [6] |
(a) A.F. Littke, G.C. Fu, Angew. Chem. Int. Ed. 41 (2002) 4176-4211; (b) P. Fitton, E.A. Rick, J. Organomet. Chem. 28 (1971) 287-291; (c) R. Stürmer, Angew. Chem. 111 (1999) 3509-3510. |
| [7] |
(a) V.V. Grushin, H. Alper, Chem. Rev. 94 (1994) 1047-1062; (b) R. Stürmer, Angew. Chem. Int. Ed. 38 (1999) 3307-3308; (c) H.U. Blaser, E. Schmidt, Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2004; (d) S.L. Buchwald, C. Mauger, G. Mignani, U. Scholz, Adv. Synth. Catal. 348 (2006) 23-39; (e) B. Schlummer, U. Scholz, Adv. Synth. Catal. 346 (2004) 1599-1626; (f) H.U. Blaser, A. Indolese, F. Naud, U. Nettekoven, A. Schnyder, Adv. Synth. Catal. 346 (2004) 1583-1598; (g) C. Torborg, M. Beller, Adv. Synth. Catal. 351 (2009) 3027-3043; (h) F. Naso, F. Babudri, G.M. Farinola, Pure Appl. Chem. 71 (1999) 1485-1492; (i) R.N. Yi, Z.J. Wang, Z.W. Liang, et al., Appl. Organomet. Chem. 33 (2019) e4917-e4923. |
| [8] |
(a) C.C.C. Johansson Seechurn, M.O. Kitching, T.J. Colacot, V. Snieckus, Angew. Chem. Int. Ed. 51 (2012) 5062-5085; (b) C.C.C. Johansson Seechurn, M.O. Kitching, T.J. Colacot, V. Snieckus, Angew. Chem. 124 (2012) 5150-5174. |
| [9] |
(a) Z.H. Jia, Liu Q, X.S. Peng, H.N.C. Wong, Nat. Commun. 7 (2016) 10614-10620; (b) C. Han, S.L. Buchwald, J. Am. Chem. Soc. 131 (2009) 7532-7533; (c) B.D. Sherry, A. Fürstner, Acc. Chem. Res. 41 (2008) 1500-1511; (d) S.M. Neumann, J.K. Kochi, J. Org. Chem. 40 (1975) 599-606; (e) A. Fürstner, A. Leitner, Angew. Chem. Int. Ed. 41 (2002) 609-612; (f) B.J. Li, L. Xu, Z.H. Wu, et al., J. Am. Chem. Soc. 131 (2009) 14656-14657; (g) C.S. Yeung, V.M. Dong, J. Am. Chem. Soc. 130 (2008) 7826-7827; (h) J. Zhou, G.C. Fu, J. Am. Chem. Soc. 125 (2003) 14726-14727; (i) K. Tamao, Y. Kiso, K. Sumitani, M. Kumada, J. Am. Chem. Soc. 94 (1972) 9268-9269. |
| [10] |
(a) D.W. Old, J.P. Wolfe, S.L. Buchwald, J. Am. Chem. Soc.120 (1998) 9722-9723; (b) J.P. Wolfe, R.A. Singer, B.H. Yang, S.L. Buchwald, J. Am. Chem. Soc.121 (1999) 9550-9561; (c) M. Schomaker, T.J. Delia, J. Org. Chem. 66 (2001) 7125-7128; (d) A.J. Cocuzza, D.R. Chidester, S. Culp, L. Fitzgerald, P. Gilligan, Bioorg. Med. Chem. Lett. 9 (1999) 1063-1066; (e) J.P. Wolfe, S.L. Buchwald, Angew. Chem. 111 (1999) 2570-2573. |
| [11] |
(a) J.M. Lovell, J.A. Joule, Synth. Commun. 27 (1997) 1209-1215; (b) A. Suzuki, J. Organomet. Chem. 576 (1999) 147-168; (c) G.B. Smith, G.C. Dezeny, D.L. Hughes, A.O. King, T.R. Verhoeven, J. Org. Chem. 59 (1994) 8151-8156. |
| [12] |
J. Zhang, J. Chen, M. Liu, X. Zheng, J. Ding, H. Wu, Green Chem. 14 (2012) 912-916. DOI:10.1039/c2gc16539b |
| [13] |
(a) K. Itami, M. Mineno, N. Muraoka, J.I. Yoshida, J. Am. Chem. Soc. 126 (2004) 11778-11779; (b) L.W. Qian, M. Sun, J. Dong, Q. Xu, Y. Zhou, S.F. Yin, J. Org. Chem. 82 (2017) 6764-6769; (c) K. Itami, D. Yamazaki, J.I. Yoshida, J. Am. Chem. Soc.126 (2004) 5396-15397; (d) S.R. Dubbaka, P. Vogel, Angew. Chem. Int. Ed. 44 (2005) 7674-7684; (e) S.R. Dubbaka, P. Vogel, Tetrahedron Lett. 47 (2006) 3345-3348; (f) P. Leowanawat, N. Zhang, A.M. Resmerita, B.M. Rosen, V. Percec, J. Org. Chem. 76 (2011) 9946-9955. |
| [14] |
(a) D. Gärtner, A.L. Stein, S. Grupe, J. Arp, A.J. Wangelin, Angew. Chem. Int. Ed. 54 (2015) 10545-10549; (b) M.R. Harris, L.E. Hanna, M.A. Greene, C.E. Moore, E.R. Jarvo, J. Am. Chem. Soc. 135 (2013) 3303-3306; (c) K.W. Quasdorf, X. Tian, N.K. Garg, J. Am. Chem. Soc. 130 (2008) 14422-14423. |
| [15] |
(a) G.G. Meng, S.C. Shi, M. Szostak, ACS Catal. 6 (2016) 7335-7339; (b) K.R. Buszek, N. Brown, Org. Lett. 9 (2007) 707-710; (c) M. Barbero, S. Dughera, Tetrahedron 74 (2018) 5758-5769; (d) Y. Bourne-Branchu, C. Gosmini, G. Danoun, Chem.-Eur. J. 25 (2019) 2663-2674. |
| [16] |
(a) H. Li, J. Liu, C.L. Sun, B.J. Li, Z.J. Shi, Org. Lett. 13 (2011) 276-279; (b) S. Tang, L. Zeng, A.W. Lei, J. Am. Chem. Soc. 140 (2018) 13128-13135; (c) Q. Zhang, Y. Liu, T. Wang, et al., J. Am. Chem. Soc. 140 (2018) 5579-5587; (d) M. Shang, H.L. Wang, S.Z. Sun, H.X. Dai, J.Q. Yu, J. Am. Chem. Soc. 136 (2014) 11590-11593. |
| [17] |
G. Booth, Nitro Compounds, Aromatic; Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, New York, 2012.
|
| [18] |
Y. Yang, Angew. Chem. Int. Ed. 56 (2017) 15802-15804. DOI:10.1002/anie.201708940 |
| [19] |
X.W. Zheng, J.C. Ding, J.X. Chen, et al., Org. Lett. 13 (2011) 1726-1729. DOI:10.1021/ol200251x |
| [20] |
J.L. Zhang, J.X. Chen, M.C. Liu, et al., Green Chem. 14 (2012) 912-916. DOI:10.1039/c2gc16539b |
| [21] |
D.P. Peng, A.J. Yu, H.L. Wang, Y.J. Wu, J.B. Chang, Tetrahedron 69 (2013) 6884-6889. DOI:10.1016/j.tet.2013.05.112 |
| [22] |
H.L. Wang, A.J. Yu, A.J. Cao, J.B. Chang, Y.J. Wu, Appl. Organomet. Chem. 27 (2013) 611-614. |
| [23] |
T. Begum, M. Mondal, M.P. Borpuzari, et al., Eur. J. Org. Chem. (2017) 3244-3248. |
| [24] |
S.S. Bahekar, A.P. Sarkate, V.M. Wadhai, P.S. Wakte, D.B. Shinde, Catal. Commun. 41 (2013) 123-125. DOI:10.1016/j.catcom.2013.07.019 |
| [25] |
H. Tian, A.J. Cao, L.J. Qiao, et al., Tetrahedron 70 (2014) 9107-9112. DOI:10.1016/j.tet.2014.09.087 |
| [26] |
A. Rostami, A. Rostami, A. Ghaderi, J. Org. Chem. 80 (2015) 8694-8704. DOI:10.1021/acs.joc.5b01248 |
| [27] |
B.P. Fors, S.L. Buchwald, J. Am. Chem. Soc. 131 (2009) 12898-12899. DOI:10.1021/ja905768k |
| [28] |
M.R. Yadav, M. Nagaoka, M. Kashihara, et al., J. Am. Chem. Soc. 139 (2017) 9423-9426. DOI:10.1021/jacs.7b03159 |
| [29] |
F. Inoue, M. Kashihara, M.R. Yadav, Y. Nakao, Angew. Chem. Int. Ed. 56 (2017) 13307-13309. DOI:10.1002/anie.201706982 |