 2019, Vol. 30
2019, Vol. 30 


Hyperbranched polypeptides that combine excellent physicochemical properties of hyperbranched polymers with good biocompatibility and degradability of polypeptides, have broad potential applications in diagnostics, drug/protein delivery and gene transfection [1]. The controlled synthesis, functionalization and self-assembly of hyperbranched polypeptides have become important topics in the fields of polymer science and biomedicine [2, 3]. For example, Klok et al. synthesized a series of hyperbranched polylysines by iterative ROP method [4]. By combining ROP with thiol-yne click chemistry, hyperbranched polylysine derivatives can be prepared in a fast manner [5]. Very recently our group developed the light/temperature triggered ROP methodologies for synthesis of hyperbranched polylysines with controlled molecular weights and degree of branching (DB) [6, 7]. However, the detailed mechanism of NH3BF4-Lys NCA ROP is unclear, and the aqueous self-assembly of hyperbranched polylysine has not been studied until now [6, 7]. Meanwhile, the biocompatible polymers stabilized gold nanoparticles (Au NPs) that exhibit excellent optical properties in near-infrared (NIR) regions have great potential applications in photothermal therapy, bioimaging, and diagnostics, in which the polymers can tune the plasmonic wavelength and enhance the stability and biocompatibility of Au NPs [8-10]. So we further tested to use as-synthesized hyperbranched polylysine as a stabilizer for synthesis of plasmonic Au NPs by utilizing multivalent Au-N coordination interactions. The as-synthesized gold nanoparticles (HPlys@Au NPs) exhibited good NIR absorption, making them promising for NIR-mediated photothermal therapy [11, 12].
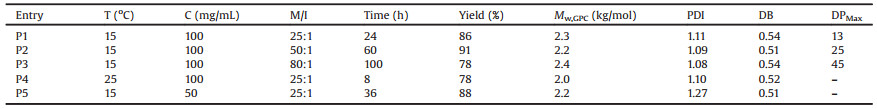
Inspired by the acid-base dynamic chemistry and organic amine salt for the controlled ROP of NCA monomers [13-16], we previously designed and synthesized an ionic monomer ε-tetrafluoroboron-L-lysine-N-carboxyanhydride (NH3BF4-Lys NCA) [7]. When dissolved in DMF above 20 ℃, it formed an active AB* type NCA (i.e., inimer-NCA) as the amino fluoroborate group dynamically dissociated; the NH3BF4-Lys NCA ROP spontaneously occurred without adding an initiator or a catalyst. The spontaneous polymerization of NH3BF4-Lys NCA is completely prohibited at 20 ℃, however, the degree of branching (DB) of the resultant hyperbranched polylysines decreased from 0.53 to 0.34 when the temperature increased from 25 ℃ to 55 ℃ [7]. As triethylamine (Et3N) can initiate NCA polymerization to produce high molecular weight polypeptides according to activated monomer mechanism (AMM), we tested to use triethylamine to accelerate ROP at a low temperature of 15 ℃ [3]. As shown in ATR-FT-IR (Fig. S1 in Supporting information), the NCA anhydride signal peaks gradually disappeared over time, demonstrating NH3BF4-Lys NCA ROP indeed occurred at 15 ℃ in the presence of Et3N. The polymerization time prolonged from 24 h to 100 h with the feed ratio of [monomer]/[Et3N] increasing from 25/1 to 80/1 (Table 1). This result is inconsistent with AMM of NCA ROP initiated by Et3N, implying that the NH3BF4-Lys NCA ROP initiated by Et3N was probably according to normal amine mechanism (NAM, Scheme 1). We used 1H-13C HSQC, NMR, and mass spectroscopy to elucidate the microstructure of the result polymers (Fig. 1). 1H-13C HSQC spectrum of P3 clearly shows three structural units: dendritic unit (D), ε-linear unit (L) and terminal unit (T), verifying its hyperbranched structure [4]. The lysine in each unit has a different chemical shift signal of methine (α-CH), and DB can be calculated by the formula DB = (D + T) / (D + T + L). Based on 1H NMR spectrum of P3, its DB is calculated as [D(1'+1) + T(3)]/[D(1'+1) + T(3) + L(4)] = 0.54 (Fig. S2 in Supporting information) [6, 7]. The Mw,GPC and DB of as-synthesized polymers did not change obviously while the maximal degree of polymerization (DP) increased, as determined by matrix-assisted laser desorption ionization time-offlight mass spectrometry (MALDI-TOF-MS, Figs. S3 and S4 in Supporting information). These results also evidence that the hyperbranched polylysine fluoroborates mainly formed with an over underestimated hydrodynamic volume in DMF.
|
|
Table 1 Synthesis of hyperbranched polylysine fluoroborates by Et3N initiated NH3BF4-Lys NCA ROP. |

|
Download:
|
| Scheme 1. The postulated routes for NH3BF4-Lys NCA ROP initiated by Et3N at 15 ℃. | |

|
Download:
|
| Fig. 1. The structure of hyperbranched polylysine (A), 1H-13C HSQC spectrum (B) and 1H NMR spectrum (C) of P3 in CD3OD. | |
The detailed microstructure of hyperbranched polymers was analyzed by MALDI-TOF-MS, as shown in Table 2 and Fig. S4. There are two possible paths for NH3BF4-Lys NCA ROP in presence of triethylamine (Scheme 1): (1) As a strongbase, Et3N neutralizes HBF4 of NH3BF4-Lys NCA and releases the inimer-NCA with an active ε-NH2, followed by the dimer cyclization and subsequent chain growth according to NAM, i.e., the cyclic dimer as a secondary initiating species, path 1; (2) As a primary initiating species, the released inimer-NCA directly initiates ROP but does not undergo cyclization for chain growth according to NAM, i.e., path 2. If the polymerization is via path 1, the charge-to-mass ratio (m/z) of the polymer corresponds to an integral number of lysine segments (128.095 Da) and cation H+ or Na+, which can be expressed as m/z = xLys + 1 (or 23) (Ⅰ). The signal peaks of A1-A13 in Table 2 all conform to formula (Ⅰ). For example, m/z of A1 = 535.370 corresponds to 4×128.095 + 22.990 = 535.370. If the polymerization is via path 2, the m/z of the polymer corresponds to an integer number of lysine segments, the terminal group 18 Da (i.e., -H + -OH, hydrolysis from the NCA ring), and the cation H+ or Na+ plus, can be expressed as m/z = xLys + 18 + 1 (or 23) (Ⅱ). The signal peaks of B1-B13 in Table 2 all conform to formula (Ⅱ). For example, m/z of B1 = 553.381 corresponds to 4×128.095 + 18.011 + 22.990 = 553.381. However, very weak signal peaks satisfying this formula are seen in the spectrum of P3. That is to say, the secondary species of cyclic dimers mainly initiated NH3BF4-Lys NCA ROP while only a small portion of primary inimer-NCA initiated ROP. By MALDI-TOF-MS analysis, it can be concluded that the fast cyclization reaction mainly occurred during the polymerization process, which consumed most of monomers, inducing low molecular weight polymer formed [6, 7]. In order to directly prove the presence of the cyclic dimer species in the polymerization, an equimolar amount of Et3N was added to accelerate the monomer dissociation at 15 ℃ (1 mg/mL, P6) and then the crude product was characterized by electrospray ionization mass spectroscopy (ESI-MS, Table S1 and Fig. S5 in Supporting information). The ESI-MS analysis indeed demonstrates that a large amount of cyclic dimer formed besides other oligomers having a cyclic dimer center and a small portion of linear oligomers.
|
|
Table 2 The microstructures of P3 analyzed by MALDI-TOF-MS. |

{kind=link}
{kind=link}
To support the point that the linear dimer is more favorable to the forming of a cyclic dimer species than initiating the third NCA monomer (i.e., the propagation reaction), we turned to the DFT computation [17-19]. The free energy barrier for the dimer cyclization reaction calculated by DFT is 15.42 kcal/mol, while the barrier for the propagation reaction is 15.93 kcal/mol. The 0.5 kcal/mol higher barrier resulted in a 2.5-fold faster cyclization reaction than propagation reaction. The calculation results indicate that the linear dimer is more likely to cyclize firstly instead of initiating the third NCA to form a linear trimer directly, that is to say, the secondary initiating species is indeed the cyclic dimer during the polymerization process.
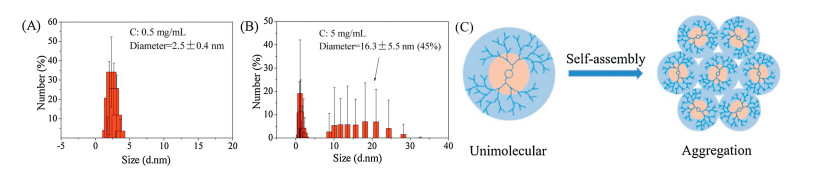
Hyperbranched polylysine (HPlys) has good water solubility at room temperature. To study whether it forms single molecular micelle or an assembled aggregate in water, we tested the HPlys aqueous solution at various concentrations of 0.5, 1, 1.5, and 5 mg/mL by using DLS. The hydrodynamic size ranged from 2.5 ± 0.4 nm at 0.5 mg/mL to 3.4 ± 0.9 nm at 1 mg/mL; however, 28% big aggregates of 8.3 ± 4.3 nm and 45% bigger ones of 16.3 ± 5.5 nm formed at 1.5 mg/mL and 5 mg/mL, respectively (Figs. 2A and B and Fig. S6 in Supporting information). As determined by TEM (Fig. S7 in Supporting information), the hyperbranched polylysine assemblies at 5 mg/mL gave spherical micellar morphology and the average nanoparticle size was about 13.2 nm, which is consistent with the DLS result. By using material studio software, the hyperbranched polylysine with a similar molecular weight of 2163 Da was simulated with a particle size of about 3 nm (Fig. S8 in Supporting information), which is in agreement with the sizes for those at 0.5 mg/mL and 1.0 mg/mL determined by DLS. Therefore, we can speculate that the hyperbranched polylysine exists as a single molecular micelle in dilute solution (≤ 1 mg/mL), and the molecular aggregation increasingly occurred over the concentration (Fig. 2C) [1-3].

|
Download:
|
| Fig. 2. DLS data of HPlys in water (A, B); Scheme for single HPlys molecule self-assembles into micellar aggregate (C). | |
{kind=link}
We speculated that the above-synthesized hyperbranched polylysine with multiple amino groups can interact and stabilize gold by the multivalent coordination of gold-nitrogen (Au-N) bondings, so we tested to prepare plasmonic Au NPs stabilized by HPlys (i.e., HPlys@Au). The HPlys aqueous solution (1 mL, 1 mg/mL) is added with a small portion of H2O2 (1 mL, 30 wt%), followed by addition of HAuCl4 (0.7 mL, 10 mg/mL), and then vigorously stirred for 24 h at room temperature in the dark. The excess polymer was removed by centrifugation for three times and the dark brown HPlys@Au NPs were dispersed in ultrapure water (Fig. 3A). WAXD showed three characteristic Bragg diffraction peaks at 38.2°, 44.3°, and 64.5°, corresponding to the (111), (200), and (220) reflections of the cubic gold phase in HPlys@Au NPs (Fig. S9 in Supporting information). As shown in Fig. 3B, the XPS of HPlys@Au NPs gave three peaks at 399.5 eV, 400.4 eV and 401.5 eV, being assigned to Au-NH2, NH3+ and NHCO, respectively [20]. This result verifies that HPlys indeed played multivalent coordination interactions for stabilizing the reduced gold nanocrystal. The DLS-determined hydrodynamic diameter for HPlys@Au NPs (Au/N ratio = 0.88) is 112.4 ±17.7 nm (Fig. 3C), which is consistent with that of about 103 nm determined by TEM (Fig. 3D). Especially, the TEM image of HPlys@Au NPs gave a high contrast with spherical gold core of about 100 nm, which was coated by a thinner polymer layer of about 3 nm. In all, the above results convincingly demonstrated the hyperbranched polylysine mainly played a stabilizer role for wrapping the gold nanocrystal [11, 12]. The Vis-NIR spectroscopy shows that Hplys@Au NPs have strong and broad absorption at 500–1100 nm and the intensity is significantly enhanced at Au/ N = 0.88, indicating good NIR absorption capacity (Fig. 3E). Upon 808 nm laser irradiation at a power intensity of 2 W/cm2, the solution temperature of Hplys@Au NPs (1 mg/mL) increased sharply over 5 min irradiation time with a maximum rise of 23.1 ℃ and a high photothermal conversion efficiency of 35.1% (Fig. S10 in Supporting information), however, the control water was elevated only by 2.3 ℃. Furthermore, the curves of repeated laser-on-off experiments are coincident, demonstrating HPlys@Au NPs with good photostability (Fig. 3F). These results evidence that the plasmonic HPlys@Au NPs have good NIR absorption and photothermal properties, enabling them potential for NIR-mediated photothermal therapy [11, 12].

|
Download:
|
| Fig. 3. The photograph (A), XPS (B), DLS (C), and TEM image (D) of HPlys@Au NPs with Au/N ratio of 0.88; (E) Vis-NIR absorption curves of HPlys@Au NPs with different Au/N ratios; (F) three repeated photothermal cycles of HPlys@Au NPs (Au/N = 0.88, 1 mg/mL) dispersed in water. | |
{kind=link}
In summary, the hyperbranched polylysine fluoroborate was prepared by NH3BF4-Lys NCA ROP in triethylamine at a low temperature of 15 ℃. The mass spectroscopy analysis and DFT computation results confirmed that the cyclic dimer mainly initiated ROP as the secondary initiating species. The hyperbranched polylysine tends to form a unimolecular micelle of 2–3 nm at ≤ 1 mg/mL but selfassembles into the micellar aggregates of about 13 nm at 5 mg/mL. The NIR-absorbing HPlys@Au NPs can be further prepared by using the hyperbranched polylysine asa stabilizer, upon NIR irradiation (808 nm, 2 W/cm2, 5 min), which attained an elevation of 23.1 ℃ with good photostability.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21474061), the Innovation Program of Shanghai Municipal Education Commission (No. 201701070002E0061), and the Innovation Fund (No. IFPM2016B004) of Shanghai Jiao Tong University & Affiliated Sixth People's Hospital South Campus are appreciated.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.03.009.
| [1] |
Y. Zheng, S. Li, C. Gao, Chem. Soc. Rev. 44 (2015) 4091-4130. DOI:10.1039/C4CS00528G |
| [2] |
Y. Huang, D. Wang, X. Zhu, D. Yan, R. Chen, Polym. Chem. 6 (2015) 2794-2812. DOI:10.1039/C5PY00144G |
| [3] |
T.J. Deming, Bioconjugate Chem. 28 (2017) 691-700. DOI:10.1021/acs.bioconjchem.6b00696 |
| [4] |
M. Scholl, T. Nguyen, B. Bruchmann, H. Klok, J. Polym. Sci. Polym. Chem. 45 (2007) 5494-5508. DOI:10.1002/(ISSN)1099-0518 |
| [5] |
X. Chang, C.M. Dong, Biomacromolecules 14 (2013) 3329-3337. DOI:10.1021/bm400951m |
| [6] |
P. Li, C.M. Dong, ACS Macro Lett. 6 (2017) 292-297. DOI:10.1021/acsmacrolett.7b00167 |
| [7] |
P. Li, C.M. Dong, Acta Polym. Sinica 1 (2018) 63-71. |
| [8] |
B. Gao, M.J. Rozin, A.R. Tao, Nanoscale 5 (2013) 5677-5691. DOI:10.1039/c3nr01091k |
| [9] |
G. Chen, I. Roy, C. Yang, P.N. Prasad, Chem. Rev. 116 (2016) 2826-2885. DOI:10.1021/acs.chemrev.5b00148 |
| [10] |
X. Yang, M. Yang, B. Pang, M. Vara, Y. Xia, Chem. Rev. 115 (2015) 10410-10488. DOI:10.1021/acs.chemrev.5b00193 |
| [11] |
J.B. Song, P. Huang, H.W. Duan, X.Y. Chen, Acc. Chem. Res. 48 (2015) 2506-2515. DOI:10.1021/acs.accounts.5b00059 |
| [12] |
X. Wu, L. Zhou, Y. Su, C.M. Dong, Biomacromolecules 17 (2016) 2489-2501. DOI:10.1021/acs.biomac.6b00721 |
| [13] |
I. Conejos-Sánchez, A. Duro-Castano, A. Birke, M. Barz, M. Vicent, J. Polym. Chem. 4 (2013) 3182-3186. DOI:10.1039/c3py00347g |
| [14] |
C.D. Vacogne, H. Schlaad, Chem. Commun. 51 (2015) 15645-15648. DOI:10.1039/C5CC06905J |
| [15] |
W. Zhao, Y. Gnanou, N. Hadjichristidis, Chem. Commun. 51 (2015) 3663-3666. DOI:10.1039/C4CC09055A |
| [16] |
J. Cheng, T.J. Deming, Peptide-Based Materials. Berlin: Springer, 2011: 1-26.
|
| [17] |
A.D. McLean, G.S. Chandler, J. Chem. Phys. 72 (1980) 5639-5648. DOI:10.1063/1.438980 |
| [18] |
Y. Zhao, D.G. Truhlar, Theor. Chem. Acc. 120 (2008) 215-241. DOI:10.1007/s00214-007-0310-x |
| [19] |
S. Sinha, R.K. Rahman, A. Raj, Phys. Chem. Chem. Phys. 19 (2017) 19262-19278. DOI:10.1039/C7CP02539D |
| [20] |
C. svoranu, B. Wang, E. Ataman, et al., J. Chem. Phys. 134 (2011) 114710. DOI:10.1063/1.3563635 |