 2019, Vol. 30
2019, Vol. 30 


b University of the Chinese Academy of Sciences, Beijing 100049, China
The organic nature of polymer active materials gives several distinct advantages, such as light weight, good mechanical flexibility, excellent solution processability [1]. Controlling bulk heterojunction (BHJ) morphology of active layers is a critical requirement for achieving high device efficiency in polymer solar cells (PSCs) [2]. The bicontinuous interpenetrating network morphology with optimal domain size and the large interfacial area is profitable for efficient devices. Recent studies have identified the morphology of BHJ structure in more detail can comprise at least three-phase network: relatively pure polymers, pure fullerenes domains, and molecularly mixed regions between polymer/fullerene [3]. The intermixed phase offers a large interfacial area between donor and acceptor aggregated domains, promoting exciton dissociation via charge separation [4]. In addition, efficient charge transport and collection in BHJ OSCs require that the donor and the acceptor materials should phase separate and crystallize appropriately while also providing continuous pathways for the transport of electrons and holes. The three-phase BHJ morphology has been demonstrated to have dramatic impacts on the device performance [4-6].
Molecular mixing of polymer donor and fullerene acceptor results in significant energy levels shift (magnitude from 100 MeV to 350 MeV) between intermixed phases and the relatively pure phases, constructing an energy cascade that assists in free charge generation [7, 8]. The function of the mixed region in promoting geminate splitting has been also studied using Kinetic Monte Carlo (KMC) simulations of idealized trilayer (pure donor/mixed region/pure acceptor) morphologies, showing that 200 MeV energetic offset between the mixed and pure regions can greatly decrease the local mobilities and CT lifetimes required for efficient charge generation [9]. The kinetic Monte Carlo (KMC) model has also demonstrated the degree of benefit of the cascade depend on the thickness of cascade layer [7]. However, complete mixing is not desirable; the coexistence of the mixed phase with relatively pure phases is necessary for efficient charge generation. Polarons in the highly mixed region are more localized and higher recombination rate than pure phases, so charges are short-lived in the mixed region [7]. In addition, intermixed regions seriously reduce the charge mobility leading to nongeminate charge recombination [10]. Pure phases are essential to prevent geminate recombination of bound electron-hole pairs [11, 12]. The presence of local aggregation can result in adequate local charge mobility to overcome the Coulombic barrier and reduce the Coulombic barrier by increasing the effective initial separation distance between electrons and holes [9, 13]. Therefore, it is of great importance for controlling the composition of pure and intermix phases to identify the relationships between specific structures and their solar cell properties.
Solution aging process is a universal method to regulate conformation of polymer in solution, such as semi-crystalline polymer P3HT, PBTTT and polymers with poor crystallinity, i.e., PTB1. For example, the interaction of PTB1 and PCBM along the direction parallel to the substrate increase after solution aging, reducing interchain d-spacing (da) and then enhanced the electron-hole separation between donor and acceptor directly [14]. For polymer/non-fullerene system, the solution aging can also be an alternative way tune polymer aggregation. Polymer aggregation in solution plays a primary role in determining the BHJ morphology.
The polymer PBTTT is a high mobilities and crystallinity liquidcrystalline material, having strong intermolecular interaction [15]. PBTTT can form a highly ordered structure due to interdigitation of side-chains between adjacent lamellae. The extended π-system geometry of PBTTT can promote lamellar packing. Alkyl side-chains are only attached to unfused thiophene rings, which permits the so-called 'intercalation' of small molecules between the extended chains [16]. Along with one [6, 6]-phenyl-C71-butyric acid methyl ester (PC71BM) molecule per PBTTT monomer (50 mol%) leads to fullerenes intercalating into polymer side chains, forming cocrystal because of electrostatic interactions between the polymer and fullerene [17]. Conjugated polymer chain aggregation in solution prior to film fabrication is critical for their film structure [18-20]. Conjugated polymer can self-assembly leads to ordered polymer aggregates with the solution aging process [21]. Considering the mixed regions are severely disordered and the vague quantitative relationship between the charge separation on the content of intermixed phase. Herein, PBTTT/PC71BM is employed as structure model for controlling and characterizing the variation of phase morphology by adjusting the extent of fullerene intercalation. Taking into account the root causes of the co-crystal intermixed phase, we utilize the solution aging to make the PBTTT polymers self-assembly in solution forming aggregates, reducing the exposure of side chain, thus manipulating the PC71BM intercalation in polymer. Controlling the solution aging time, we have obtained microstructures with different contents of intermixed phases. As the content of intercalated phase is about 11%, the charge dissociation is most efficiently and short circuit current (Jsc) optimal. Our findings show that three-phases morphologies comprised predominant pure phases and little mixed phase are more efficient for exciton dissociation, thus effective for charge photogeneration.
Polymer aggregation in solution plays a significant role in determining the morphology [22]. To characterize the conformation and aggregation of the polymer PBTTT in solution along with aging time, we examined the absorption of PBTTT solutions with ultraviolet-visible (UV–vis) spectroscopy, as shown in Fig. 1. The absorption spectrum of PBTTT in solution is characterized by two spectral features, a high-energy peak attributed to the π-π* transition with a peak at 590 nm and a strong absorption peak at 480 nm. The max absorption peak intensity gradually decreased and shifted to slightly higher wavelength upon aging due to selforganization of polymers results in changes of optical density [23]. The intensity of π-π stacking peak at 590 nm was increased with extending the aging time which indicated the molecules were aggregated. This finding suggests that the content of PBTTT chains that are aggregated in solution increases as the aging time is increasing.

|
Download:
|
| Fig. 1. The UV–vis absorption spectra for PBTTT solution with different aging time. | |
{kind=link}
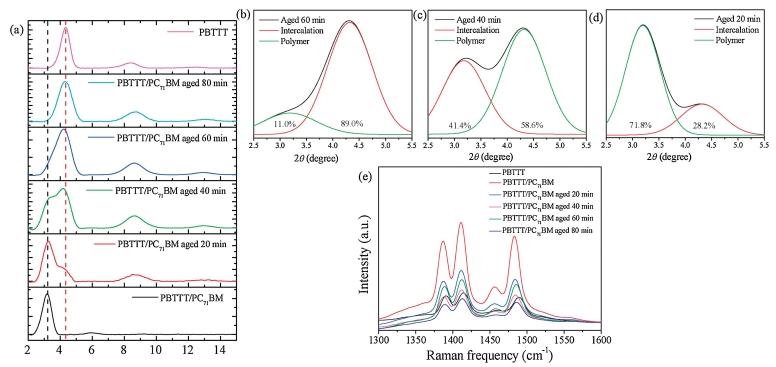
We utilized ordered aggregates of PBTTT chains via solution aging process to manipulate the PC71BM intercalation extent in the PBTTT polymer; ie controlling the amount of this ordered intermixed phase. Therewith we can tune the ratio between mixed and pure phase in PBTTT/PC71BM system to investigate the effects of an altered microstructure on charge carrier photogeneration. To verify the PC71BM intercalation extent in the PBTTT polymer in the PBTTT/PC71BM blend film spin-coated from without and with aged solutions, out-of-plane GIXD patterns had been performed, the results are present in Fig. 2a. The (100) diffraction peak of neat PBTTT film is at 2θ = 4.35°, leading to a repeat distance d100 of 20.3 Å. Blending PC71BM with PBTTT increases this spacing to 27.8 Å (2θ = 3.17°) in the PBTTT/PC71BM blend due to fullerene intercalation between the interdigitated side chains of the polymer. Such an expansion of lattice in the d spacing of 7.5 Å is in good agreement with the values published in the literature [24]. Solution aging process promotes the formation of polymer order aggregates in solution, making fractions of PC71BM do not intercalate during formation of the blend film. This leads to an amount of relatively pure PBTTT region. When the solution aged 20 min, we observe the diffraction peak is not symmetrical due to the formation of neat polymer phase. The strong diffraction observed at 2θ = 3.17° indicates that an intercalated phase predominates in three phases. Through peak fitting for separating the peak of intercalation and polymer, the calculated content of neat polymer phase is 28.2%. Along with the aged time, more fractions of PC71BM do not intercalate in the aged 40 min and 60 min cases, leading to a higher fraction of phase-pure PBTTT domains, as evident from the diffraction observed at 2θ = 4.35°. The calculated content of neat polymer phase is 58.6% and 89%, respectively. As the solution aged 80 min, the polymer aggregated seriously making PC71BM intercalate with difficulty, as consequence of a nearly two-phase comprising pure PBTTT and PC71BM formed. Thus, regulating the polymer solution aggregation, we can induce either a fully intercalated, partially intercalated, or predominantly non-intercalated system.
These findings are supported by Raman spectroscopy measurements, as shown in Fig. 2e. Compared to the pristine PBTTT, Raman spectra of PBTTT/PC71BM are redshifted notably which is consistent with the co-crystal formation. Meanwhile, Raman transitions from PC71BM are observed apparently with dominant C=C breathing mode arising at ≈1460 cm-1 [25]. Therefore, the 1460 cm-1 Raman intensity and the red-shift extent of Raman spectra can be utilized to monitor the extent of fullerene intercalation in the polymer. The red-shift extent and the 1460 cm-1 intensity of Raman spectra of PBTTT/PC71BM cast from different aged time get smaller prolonging the aged times compared to the pure PBTTT. This implies the extent of fullerene intercalation in the polymer. As the aged time prolong, the fraction of neat PBTTT get higher, with the red-shift extent getting smaller as shown in Fig. 2e.

|
Download:
|
| Fig. 2. (a) GIXD patterns acquired from PBTTT/PC71BM blend films cast from different aged times. Peak fitting for separating the peak of intercalation and polymer (b) aged 60 min (c) aged 40 min (d) aged 20 min. (e) Raman spectra of pristine PBTTT and PBTTT/PC71BM with different aged times. | |
{kind=link}
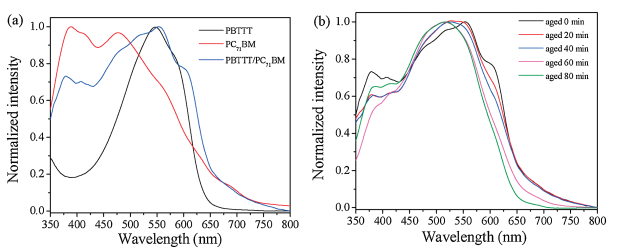
Optical absorption spectroscopy measurement was used to further study how the solution aging process influences the cocrystal structure and their ordering. As Fig. 3a shows, the absorption spectra of neat PBTTT has visible absorption peaks at 540 nm and 605 nm respectively, while neat PC71BM films absorb little in the visible range. The polymer absorption spectrum shows a more distinct absorption peak at 605 nm due to the vibronic progression and is more red-shifted when blended with PC71BM attributed to significant changes in the crystal structure between neat and intercalated PBTTT. This spectral difference between neat and intercalated PBTTT arise from PC71BM intercalation changing the backbone conformation, chain stacking, and electronic coupling of the polymer [26]. For simples cast from aged 20 min, 40 min, and 60 min solutions, the superposed absorption of the broad and structured PBTTT features shows that the PC71BM deviate from the polymer (partial intercalation structure). The absorption spectra of the PBTTT/PC71BM blends processed with the 80 min aged solution shows a structureless signature around the 540 nm and no shoulder at 605 nm, indicating primarily presence of pure PBTTT phase (predominantly none-intercalated) as shown in Fig. 3b. Meanwhile we also observe the absorption spectrum of the PBTTT/PC71BM blends are blue shifted along the aged time, confirming more and more the presence of pure phase along the aged time.

|
Download:
|
| Fig. 3. Steady-state absorption spectra of the investigated PBTTT, PC71BM, and PBTTT/PC71BM systems. (a) Neat films of PBTTT and PC71BM. (b) PBTTT/PC71BM blends spincoated from solutions with different aged time. | |
{kind=link}
Transmission electron microscopy was utilized to investigate nanoscale morphologies of PBTTT/PC71BM blends film cast from the solution aged for different periods of time. Fig. 4 shows the transmission electron microscopy images of 1:1 PBTTT/PC71BM blends with different solution aging times. In the TEM images, the dark areas are attributed to PC71BM-rich domains because the electron scattering density of PC71BM is much higher than that of PBTTT. As shown in Fig. 4a, the phase separation structure could not be observed for the as-spun film. We observed that changes of the phase separation of the film deposited from solutions aged for 20 min and 40 min became obvious. When the solution aging time increases to 60 min, the degree of phase separation enhanced and the interpenetrating networks became clearer. The rod-like fiber structures were also could be seen, which perhaps was a benefit to device performance. The film cast from a solution aged for 80 min, the phase separation became larger respect to shorter aging times.

|
Download:
|
| Fig. 4. TEM images of PBTTT/PC71BM blend films prepared by using solutions with different aged times: (a) 0 min; (b) 20 min; (c) 40 min; (d) 60 min; (e) 80 min. | |
{kind=link}
To further understand the extent of phase separation in the samples prepared here, photoluminescence (PL) spectroscopy had been conducted. The PL spectra of polymer/PC71BM blend samples with different aged time were collected, as in Fig. S2 (Supporting information). After the solutions aged for different time, we found that blend films cast from the aged solutions caused increased PL intensity. The PL intensity increases gradually along with the increase of aging time. Therefore, As shown in phase image Fig. S2b, phase separation size is directly related to the aging time. The PL results are consistent with TEM observations discussed above and our proposed morphology evolution and their mixed phases (co-crystal) in these samples.
Morphology phase separation has a great effect on the charge generation and recombination dynamics for bulk heterojunction photovoltaic blend films. For the 1:1 PBTTT/PC71BM system, PBTTT is capable of hosting fullerene between its side chains to form a known co-crystal structure with little phase-pure domains forming. It is difficult to realize the interpenetrating network structure. After solution aging process, the polymer aggregate into ordered structure, which inhibit intercalation behavior and induce phase separation.
After manipulating the extent of PC71BM intercalation and characterizing the morphology of these samples, we then fabricated photovoltaic devices to test whether the different morphologies affects the performance of devices. The photovoltaic performance data including short-circuit current (Jsc), open-circuit voltage (Voc), fill factor (FF), and power conversion efficiency values were determined (Table S1 in Supporting information). All the devices showed typical rectifying curves under 100 mW/cm2 illumination, as shown Fig. 5a.

|
Download:
|
| Fig. 5. (a) J-V curves for devices processed from PBTTT/PC71BM films with different aged time. (b) EQE of the same devices. (c) Cyclic voltammetry measurements of thin films of pure PBTTT and PBTTT/PC71BM. (d) Schematic of a BHJ solar cell including the mixed region. | |
{kind=link}
Devices made with just prepared polymer blends (0 min) showed PCE of 0.44% obtained with a Voc of 590 mV, a Jsc of 1.60 mA/cm2, and an FF of 47%. The finely intermixed texture shows poor OPV performance due to the absence of pure phases, which leads to nongeminate recombination in transport preventing extraction then poor photocurrent density. When the blend mixture is aged 20 min, the Jsc increased to 3.1 mA/cm2 and Voc slightly decreased to 460 mV, respectively. As a result, the PCE improved to 0.65%. Device shows a slight improvement in OPV performance, mainly due to the improved short-circuit current which is caused by the presence of pure PC71BM and polymer domains thus assisting in free charge generation. After aging the solution for 40 min, the Jsc improved to 3.8 mA/cm2 due to more fractions of pure polymer and PC71BM presence, while maintaining the Voc and the FF little changed. Consequently, the PCE of the PSC reached 0.77%. When the blend solution is aged for 60 min, the Jsc of the PSCs increased to 4.94 mA/cm2, thus achieving the PCE values to 1.04%. The optimum performance is obtained in the aging 60 min case. Based on our GIXD, Raman, PL and TEM data, the microstructure of the film spin-coated with the solution aging for 60 min includes a relatively large fraction of pure polymer and fullerene regions with about 11% content of intermix phase(cocrystal) being minority phases. The film architecture has optimized phase-separated morphology. Our work shows that large amounts of pure phase domains coexisting with a few mixed phases are most efficient. Further increased the solution aged time to 80 min, the PCE value dropped to 0.80%. The PCE decay is mainly due to the loss of Jsc. Aging the solution for 80 min leading to a pronounced phase-segregation of the PBTTT and PC71BM seem to be somewhat limited by exciton diffusion.
To investigate the Jsc variation, the external quantum efficiency (EQE) spectrum was used to study the spectra response of the devices for each blend aging time. The EQE curves cover a broad wavelength range from 300 nm to 750 nm as shown in Fig. 5b. The EQE is enhanced over all wavelengths with increasing blend aging times, in good agreement with the Jsc results from the current– voltage characterization of our devices. The EQE value of the devices fabricated with the aged 80 min solution was lower compared to the devices processed with the aged 60 min solution, mainly due to the two-phase structure limiting exciton diffusion to the disadvantage of free charge generation. For the PBTTT/PC71BM blend films, enhanced EQE and Jsc originate from the efficient charge dissociation and transport due to the aging process making the presence of relatively pure PBTTT and PC71BM phases assisting the co-crystal in spatial separation of photogenerated electrons and holes.
Cyclic voltammetry is an effective and powerful tool for studying energetic landscape of polymer/fullerene bulk heterojunctions [27]. Therefore, we utilized CV to measure the energy level shifts of 1:1 PBTTT/PC71BM intermixed phase and neat PBTTT, shown in image Fig. 5c. Addition of PC71BM to PBTTT has greatly impact on the PBTTT oxidation features, with higher potentials and broadened peaks compare to the pure PBTTT. We found ΔE between the PBTTT/PC71BM intermixed phase and neat PBTTT be between 260 MeV (shift of first peak) to 310 MeV (shift of second peak), respectively. As shown in Fig. 5d, the three-phase bulk heterojunction presented in these thin-film facilitated the formation of energy cascade between the mixed and pure phases, thus assisting in improving charge separation and reduce recombination.
In summary, for PBTTT/PC71BM blends, using solution aging process to make the polymer self-assembled, hindering the intercalation of PC71BM. Therefore, we can control the extent of intercalation of PC71BM in the polymer. We have realized morphologies with almost complete intercalated phase (aging 0 min)-one phase, predominant intercalated phase co-existing minority pure PBTTT and PC71BM phases (aging 20 min)-three phases, predominant pure phases with minimal intercalated phases (aging 40 min and aging 60 min) and almost nonintercalated (aging 80 min)-two phases. The optimum performance is achieved for PBTTT/PC71BM active layer spin-coated from the solution aging for 60 min, indicating the optimized morphology comprising predominant pure phases with 11% content of mixed phase. The energy cascade between the mixed and phase-pure domains is sufficiently to efficiently separate charge pairs and suppress charge recombination, so free charges subsequently percolate through the pure phase domains. Hence, balancing intermixed and pure phases for three-phase morphology should play important role in improving the device performance.
AcknowledgmentsThis work was partly supported by the National Natural Science Foundation of China (Nos. 5189071, 91833306, 51303177), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB12020300).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2019.04.004.
| [1] |
Z.H. Du, X.H. Yu, Y.C. Han, Chin. Chem. Lett. 29 (2018) 399-404. DOI:10.1016/j.cclet.2017.09.031 |
| [2] |
F.W. Zhao, C.R. Wang, X.W. Zhan, v. Ad, Energy Mater. 8 (2018) 34. |
| [3] |
M. Pfannmoller, W. Kowalsky, R.R. Schroder, Energy Environ. Sci. 6 (2013) 2871-2891. DOI:10.1039/c3ee41773e |
| [4] |
P. Westacott, J.R. Tumbleston, S. Shoaee, et al., Energy Environ. Sci. 6 (2013) 2756-2764. DOI:10.1039/c3ee41821a |
| [5] |
J.T. Bloking, T. Giovenzana, A.T. Higgs, et al., Adv. Energy Mater. 4 (2014) 12. |
| [6] |
H. Yan, Y. Song, G.R. McKeown, et al., Adv. Mater. 27 (2015) 3484-3491. DOI:10.1002/adma.201501065 |
| [7] |
C. Groves, Energy Environ. Sci. 6 (2013) 1546-1551. DOI:10.1039/c3ee24455e |
| [8] |
S. Sweetnam, K.R. Graham, G.O.N. Ndjawa, et al., J. Am. Chem. Soc. 136 (2014) 14078-14088. DOI:10.1021/ja505463r |
| [9] |
T.M. Burke, M.D. McGehee, Adv. Mater. 26 (2014) 1923-1928. DOI:10.1002/adma.201304241 |
| [10] |
G.K. Long, B. Wu, A. Solanki, et al., Adv. Energy Mater. 6 (2016) 11. |
| [11] |
F.C. Jamieson, E.B. Domingo, McCarthy-Ward T., et al., Chem. Sci. 3 (2012) 485-492. DOI:10.1039/C1SC00674F |
| [12] |
A. Zusan, K. Vandewal, B. Allendorf, et al., Energy Mater. 4 (2014) 7. |
| [13] |
H.M. Feier, O.G. Reid, N.A. Pace, et al., Adv. Energy Mater. 6 (2016) 9. |
| [14] |
P. Han, V.S. Balderrama, A. Mihi, et al., IEEE J. Photovolt. 5 (2015) 889-896. DOI:10.1109/JPHOTOV.2015.2402433 |
| [15] |
P. Brocorens, A. Van Vooren, M.L. Chabinyc, et al., Adv. Mater. 21 (2009) 1193-1198. DOI:10.1002/adma.200801668 |
| [16] |
M.L. Chabinyc, M.F. Toney, R.J. Kline, et al., J. Am. Chem. Soc. 129 (2007) 3226-3237. DOI:10.1021/ja0670714 |
| [17] |
A.C. Mayer, M.F. Toney, S.R. Scully, et al., Adv. Funct. Mater. 19 (2009) 1173-1179. DOI:10.1002/adfm.200801684 |
| [18] |
X. Gao, J.G. Liu, Y. Sun, et al., Chin. Chem. Lett. 24 (2013) 23-27. DOI:10.1016/j.cclet.2013.01.014 |
| [19] |
X.X. Cao, Z.H. Du, L. Chen, et al., Polymer 118 (2017) 135-142. DOI:10.1016/j.polymer.2017.04.076 |
| [20] |
S.J. Chi, L. Chen, J.G. Liu, et al., Chin. Chem. Lett. 28 (2017) 333-337. DOI:10.1016/j.cclet.2016.09.005 |
| [21] |
L. Chen, K.F. Zhao, X.X. Cao, et al., Polymer 149 (2018) 23-29. DOI:10.1016/j.polymer.2018.06.068 |
| [22] |
J.A. Bartelt, J.D. Douglas, W.R. Mateker, et al., Energy Mater. 4 (2014) 11. |
| [23] |
R.H. Lohwasser, M. Thelakkat, Macromolecules 44 (2011) 3388-3397. DOI:10.1021/ma200119s |
| [24] |
N.C. Cates, R. Gysel, Z. Beiley, et al., Nano Lett. 9 (2009) 4153-4157. DOI:10.1021/nl9023808 |
| [25] |
J. Gao, A.K. Thomas, R. Johnson, et al., Chem. Mater. 26 (2014) 4395-4404. DOI:10.1021/cm501252y |
| [26] |
M. Scarongella, J. De Jonghe-Risse, E. Buchaca-Domingo, et al., J. Am. Chem. Soc. 137 (2015) 2908-2918. DOI:10.1021/ja510032x |
| [27] |
S. Sweetnam, K.R. Graham, G.O.N. Ndjawa, et al., J. Am. Chem. Soc. 136 (2014) 14078-14088. DOI:10.1021/ja505463r |