 2019, Vol. 30
2019, Vol. 30 


Indoles, as important building blocks in organic synthesis, are commonly encountered in natural products, pharmaceutical molecules and functional materials [1], such as anti-Alzheimer's disease, imaging agent for protein misfolding, inhibitor of the plasmodium falciparum and cell necrosis (Fig. 1) [2]. Consequently, developing mild and efficient access to indole motif is of great significance. Organic chemists have been making great efforts to effectively synthesize indoles [3].

|
Download:
|
| Fig. 1. Selected examples illustrating the importance of the compounds with indole. | |
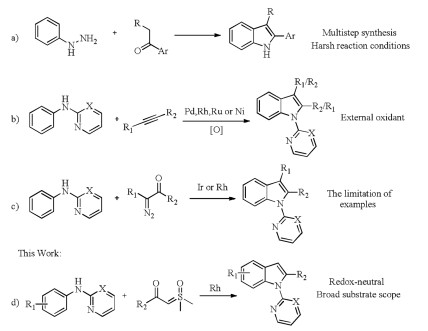
Conventional pathway involves the classical Fischer indole syntheses (Scheme 1a) [4], which suffer from tedious procedures, low atomic economy and relatively harsh reaction conditions. Recently, transition-metal-catalyzed direct C-H bond activation has emerged as a powerful tool for the synthesis of indole and its derivatives. In this context, indoles have been prepared from anilines by oxidative alkyne annulation via C-H/N-H bond functionalization catalyzed by transition-metals, such as palladium, rhodium, ruthenium and nickel (Scheme 1b) [5]. Unfortunately, these procedures were usually limited in regioselectivity when using asymmetric alkynes as substrates and relied on the external oxidants. Kim, Wang, Li and Yao groups independently reported internal molecular cyclization of diazo compounds with anilines to access indoles catalyzed by Rh and Ir under redoxneutral conditions (Scheme 1c) [6]. Despite these approaches are highly efficient and step economic, the application of diazo compounds still remains limited, especially mono-substituted diazo compounds. Subsequently, Jana group demonstrated a ruthenium(Ⅱ)-catalyzed steric controlled synthesis of 2-methylindole from anilines and allyl acetate [7]. Very recently, our group developed a palladium-catalyzed efficient cyclization reaction of anilines with vinyl azides [8], which delivered a straightforward approach to 2-arylindole derivatives with excellent regioselectivity and overcame the problem of chemo- and region-selectivity. In the meantime, sulfoxonium ylide has been developed as a versatile carbene source owing to its considerable higher security and being easily handled in comparison with diazo compounds and azides [9]. To further explore and enrich annulative synthesis of indoles, we herein proposed a protocol to various 2-substituted indoles starting from anilines using sulfoxonium ylides as carbene precursors through Rh(Ⅲ) catalyzed a C-H alkylationnucleophilic cyclization cascade reactions (Scheme 1d). During preparation of this manuscript, Huang reported a similar work [10]. Compared to Huang's work, this method is advantageous with higher yield, less amount of additive and avoiding an inert gas protection.

|
Download:
|
| Scheme 1. Synthetic strategies toward indole derivatives. | |
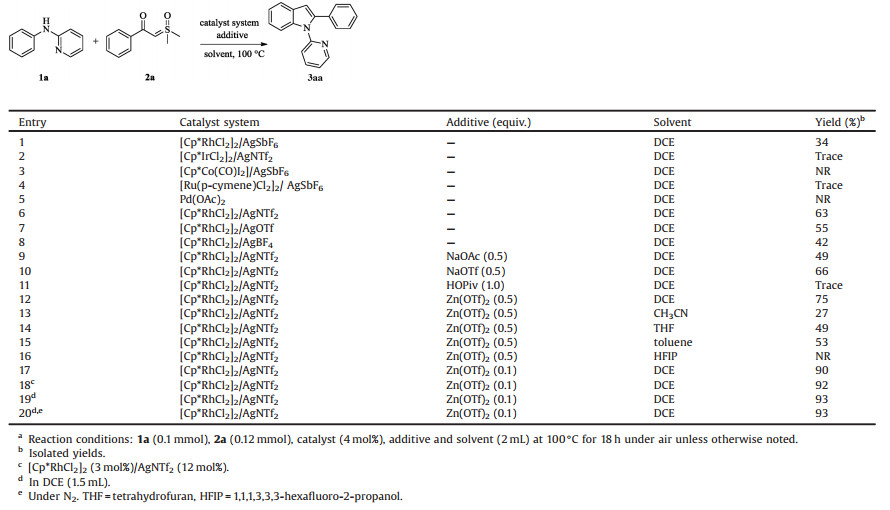
Initially, N-phenylpyridin-2-amine 1a and sulfoxonium ylide 2a were chosen as model substrates to optimize the reaction conditions (Table 1). By treating 1a (0.1 mmol) and 2a (0.12 mmol) in the presence of [Cp*RhCl2]2 (4 mol%) and AgSbF6 (16 mol%) in DCE at 100 ℃ under air, the target product 3aa was isolated in 34% yield (entry 1). The structure of 3aa was confirmed by singlecrystal X-ray diffraction analyses (CCDC 1874504). No desired product was detected using CoⅢ, IrⅢ, RuⅡ and PdⅡ as catalysts (entries 2-5). To our delight, the yield of 3aa was dramatically improved to 63% when AgNTf2 was used (entries 6-8). Subsequently, a variety of additives including NaOAc, NaOTf, HOPiv and Zn(OTf)2 were evaluated in the presence of [Cp*RhCl2]2/AgNTf2 catalyst system, and Zn(OTf)2 was shown to be the most efficient additive (entries 9-12) [11]. Encouraged by these promising data, other solvents were screened under the identical reaction conditions, and the DCE solvent proved to be superior to others, such as CH3CN, THF, toluene and HFIP (entries 13-16). Surprisingly, decreasing the loading of Zn(OTf)2 to 10 mol% led to increase of the yield of 3aa (90%, entry 17). Further studies of different catalyst loadings revealed that only 3 mol% of catalyst system provided the best yield (entry 18). When the solvent was reduced to 1.5 mL, the desired product was obtained in 93% yield (entry 19). In addition, the similar result was given under N2. Finally, the optimal reaction conditions were identified as follows: N-phenylpyridin-2-amine (0.1 mmol), sulfoxonium ylide (0.12 mmol) in the presence of [Cp*RhCl2]2 (3 mol%)/AgNTf2 (12 mol%) with Zn(OTf)2 (10 mol%) in DCE (1.5 mL) at 100 ℃ under air for 18 h.
|
|
Table 1 Optimization of the reaction conditions.a |
{kind=link}
{kind=link}
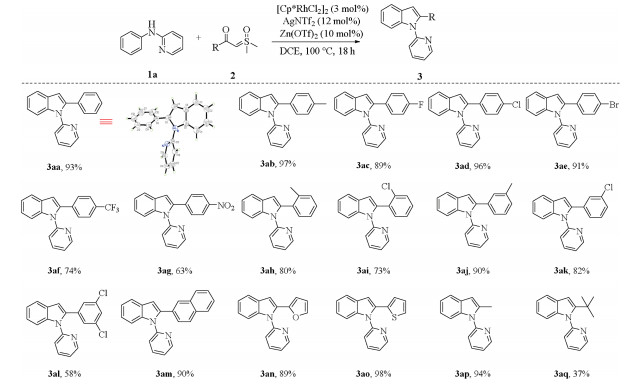
With the optimized reaction conditions in hand, we first explored the reactivity of various sulfoxonium ylides with N-2-pyridyl-substituted aniline (1a). As shown in Scheme 2, substituted benzoyl sulfoxonium ylides bearing donating group (4-methyl), withdrawing groups (4-CF3 and 4-NO2) and halogens (4-F, 4-Cl and 4-Br) all coupled smoothly with N-phenylpyridin-2-amine to deliver the corresponding products in good to excellent yields (3ab-3ag). In addition, ortho- and meta-substituted benzoyl sulfoxonium ylides were also suitable for this reaction, affording the corresponding products in high yields (3ah-3ak). Disubstituted benzoyl sulfoxonium ylide (3al) was coupled with 2a to provide indole in 58% yield. Meanwhile, this transformation also displayed an excellent tolerance toward sulfoxonium ylides including naphthyl and heterocycles (3am-3ao). More importantly, methyl and tert-butyl-substituted sulfoxonium ylides afforded the corresponding products 3ap and 3aq in 94% and 37% yields, respectively. tert-Butyl group showed poor reactivity perhaps due to its steric hindrance [12].

|
Download:
|
| Scheme 2. Scope of sulfoxoinum ylides. Reaction conditions: 1a (0.1 mmol), 2 (0.12 mmol), [Cp*RhCl2]2 (3 mol%)/AgNTf2 (12 mol%), Zn(OTf)2 (10 mol%), DCE(1.5 mL), 100 ℃, 18 h. Isolated yields. | |
{kind=link}
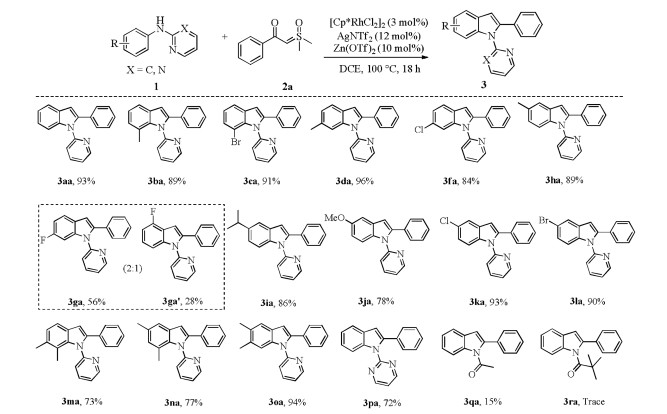
Next, the scope of N-arylpyridin-2-amines was investigated as shown in Scheme 3. It was found that N-2-pyridyl-substituted anilines with electron-withdrawing or electron-donating groups at diverse positions of the aromatic ring all underwent smoothly to afford the corresponding products in good to excellent yields (3ba-3fa, 3ha-3fa). These substituents have no significant effect on this reaction. Moreover, disubstituted N-phenylpyridin-2-amines also gave high yield (3ma-3oa). Moderate regioselectivity of 3ga:3ga' (2:1) was detected for the substrate 2g, and the major coupling product 3ga corresponds to C-C coupling at a less hindered position [5g, 8, 13]. In addition, directing groups on the N-atom of anilines, such as acetyl and pivaloyl showed less reactivity under the optimized reaction conditions (3qa and 3ra). N-Pyrimidinyl aniline was also found to be a good substrate for this annulation reaction to afford corresponding product 3pa in 72% [14].

|
Download:
|
| Scheme 3. Substrate scope of N-substituted amines. Reaction conditions: 1 (0.1 mmol), 2a (0.12 mmol), [Cp*RhCl2]2 (3 mol%)/AgNTf2 (12 mol%), Zn(OTf)2 (10 mol%), DCE (1.5 mL) at 100 ℃ for 18 h. Isolated yields. | |
{kind=link}
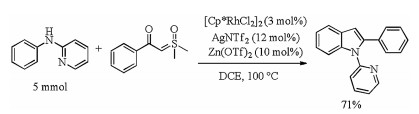
To show the utility of this catalytic system, a gram-scale synthesis of 3aa was performed under standard reaction condition with 71% yield (Scheme 4).

|
Download:
|
| Scheme 4. Large-scale synthesis. | |
{kind=link}
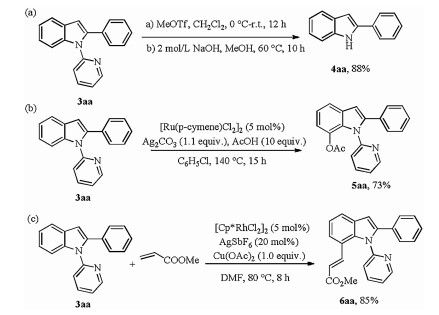
Next, the deprotection of the pyridyl group of 3aa was carried out under certain conditions, affording free N-H indole derivatives in 88% yield (Scheme 5a) [15]. Subsequently, considering the privileged structural features and biological activities of N-pyridyl-2-arylindoles, we attempted to develop the direct C-H functionalization at the C7-position. As shown in Scheme 5, the acetoxylation and alkenylation of the obtained product delivered the corresponding products catalyzed by Ru (Ⅱ)/Rh(Ⅲ) in good yields respectively (Schemes 5b and c) [16, 17]. These transformations further confirmed the practicability of the protocol.

|
Download:
|
| Scheme 5. Chemical transformation of the product. | |
{kind=link}
To better understand the reaction mechanism, several control experiments were carried out (Scheme 6). Initially, when performing the H/D exchange of N-phenylpyridin-2-amine (1a) with CD3OD under standard condition in the absence of sulfoxonium ylide (2a), incorporation (17% D) at the ortho-position of 1a was observed, suggesting the reversibility of ortho-metalation step (Scheme 6a). Next, the kinetic isotope effect via intermolecular competition reaction of 1a and 1a-d5 with 2a was investigated under standard reaction condition and kH/kD = 1.2 was obtained, which indicates that the C-H bond cleavage may not be involved in the rate-determining step (Scheme 6b). Finally, intermolecular competition experiments with differently substituted anilines displayed electron-withdrawing aniline (1k) to be converted preferentially (Scheme 6c).

|
Download:
|
| Scheme 6. Control experiments. | |
{kind=link}
Based on the control experiments and literature reports [9c, 11a, 18], a plausible reaction mechanism is proposed (Scheme 7). An active Rh(Ⅲ) complex A is generated from the reaction of aniline and [Cp*RhCl2]2 in the presence of AgNTf2 or Zn(OTf)2 via the direct ortho-metalation process. Subsequent nucleophilic attack of sulfoxonium ylide on Rh(Ⅲ) complex A gives the intermediate B, which successively undergoes the α-elimination of DMSO to produce a Rh(V) carbene C. Then migratory insertion affords the six-membered rhodacyclic intermediate D. The protondemetalation of species D delivers ortho-alkylate E and regenerates the active Rh(Ⅲ) complex for the next catalytic cycle. Finally, the desired product is obtained by an intramolecular nucleophilic attack and the release of H2O.

|
Download:
|
| Scheme 7. Plausible catalytic cycle. | |
{kind=link}
In conclusion, we have realized a rhodium(Ⅲ)-catalyzed redoxneutral coupling of N-functionalized anilines with sulfoxonium ylides, which provides an efficient method for the synthesis of indole derivatives. Mechanistically, the indoles are obtained through a C-H alkylation - nucleophilic cyclization cascade. Moreover, this transformation could smoothly proceed with high yields, good regioselectivity, no external oxidants, broad substrate scope and excellent functional group tolerance.
AcknowledgmentsWe greatly acknowledge partial financial support from the Ministry of Science and Technology of China (No. 2016YFE0132600), and Zhengzhou University.
Appendix A. Supplementary dataSupplementary material related to this article canbe found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.01.033.
| [1] |
(a) L. Li, Z. Chen, X. Zhang, Y. Jia, Chem. Rev. 118 (2018) 3752-3832; (b) L. Meng, Q. Guo, M. Chen, et al., Chin. Chem. Lett. 29 (2018) 1257-1260; (c) M. Corsello, J. Kim, N. Garg, Chem. Sci. 8 (2017) 5836-5844; (d) E. Stempel, T. Gaich, Acc. Chem. Res. 49 (2016) 2390-2402; (e) R.D. Taylor, M. MacCoss, A.D.G. Lawson, J. Med. Chem. 57 (2014) 5845-5859; (f) D.A. Horton, G.T. Bourne, M.L. Smythe, Chem. Rev. 103 (2003) 893-930. |
| [2] |
(a) J. Manfredi, U.S. Pat. Appl. Publ., 20130261118, 03 Oct. 2013; (b) I.I. Zaitseva, S.V. Zaitsev, P.O. Berggren, Invest. New. Drug. 34 (2016) 522-529; (d) L.S. Ross, M.J. Lafuente-Monasterio, T. Sakata-Kato, et al., ACS Infect. Dis. 4 (2018) 508-515; (f) D. Marie-Thérèse, B. Stéphane, D. Claire, et al., Eur. Pat. Appl. Publ. (2017) WO 2017064216, 20 Apr. |
| [3] |
(a) Z. Wang, P. Xie, Y. Xia, Chin. Chem. Lett. 29 (2018) 47-53; (b) Z. Chen, X. Shi, D. Ge, et al., Chin. Chem. Lett. 28 (2017) 231-234; (c) B. Zhang, A. Studer, Chem. Soc. Rev. 44 (2015) 3505-3521; (d) G.R. Humphrey, J.T. Kuethe, Chem. Rev. 106 (2006) 2875-2911. |
| [4] |
(a) E. Fischer, F. Jourdan, Ber. Dtsch. Chem. Ges. 16 (1883) 2241-2245; (b) E. Fischer, O. Hess, Ber. Dtsch. Chem. Ges. 17 (1884) 559-568. |
| [5] |
(a) X. Hu, X. Chen, Y. Zhu, et al., Org. Lett. 19 (2017) 3474-3477; (b) P.V. Reddy, M. Annapurna, P. Srinivas, et al., New J. Chem. 39 (2015) 3399-3404; (c) J. Chen, L. He, K. Natte, et al., Adv. Synth. Catal. 356 (2014) 2955-2959; (d) W. Song, L. Ackermann, Chem. Commun. 49 (2013) 6638-6640; (e) L. Ackermann, A.V. Lygin, Org. Lett. 14 (2012) 764-767; (f) J. Chen, Q. Pang, Y. Sun, et al., J. Org. Chem. 76 (2011) 3523-3526; (g) J. Chen, G. Song, C.L. Pan, X. Li, Org. Lett. 12 (2010) 5426-5429; (h) F. Maassarani, M. Pfeffer, J. Spencer, E. Wehman, J. Organomet. Chem. 466 (1994) 265-271. |
| [6] |
(a) K. Yu, Y. Liang, B. Li, et al., Adv. Synth. Catal. 358 (2016) 661-666; (b) H. Jiang, S. Gao, J. Xu, et al., Adv. Synth. Catal. 358 (2016) 188-194; (c) G.D. Tang, C.L. Pan, X. Li, Org. Chem. Front. 3 (2015) 87-90; (d) N. KumaráMishra, S. HoonáHan, I. SuáKim, Chem. Commun. 51 (2015) 17229-17232. |
| [7] |
M.K. Manna, G. Bairy, R. Jana, J. Org. Chem. 83 (2018) 8390-8400. DOI:10.1021/acs.joc.8b01034 |
| [8] |
L. Jie, L. Wang, D. Xiong, et al., J. Org. Chem. 83 (2018) 10974-10984. DOI:10.1021/acs.joc.8b01618 |
| [9] |
(a) C. You, C. Pi, Y. Wu, X. Cui, Adv. Synth. Catal. 360 (2018) 4068-4072; (b) G. Chen, X. Zhang, R. Jia, et al., Adv. Synth. Catal. 360 (2018) 3781-3787; (c) P. Hu, Y. Zhang, Y. Xu, et al., Org. Lett. 20 (2018) 2160-2163; (d) H. Oh, S. Han, A.K. Pandey, et al., J. Org. Chem. 83 (2018) 4070-4077; (e) X. Wu, H. Xiong, S. Sun, et al., Org. Lett. 20 (2018) 1396-1399; (f) K.S. Halskov, M.R. Witten, G.L. Hoang, et al., Org. Lett. 20 (2018) 2464-2467; (g) M. Barday, C. Janot, N.R. Halcovitch, et al., Angew. Chem. Int. Ed. 129 (2017) 13297-13301; (h) J. Vaitla, A. Bayer, K.H. Hopmann, Angew. Chem. Int. Ed. 56 (2017) 4277-4281. |
| [10] |
X. Cui, Z. Ban, W. Tian, et al., Org. Biomol. Chem. 17 (2019) 240-243. DOI:10.1039/C8OB02818D |
| [11] |
(a) Y. Xu, G. Zheng, X. Yang, et al., Chem. Commun. 54 (2018) 670-673; (b) G. Zheng, M. Tian, Y. Xu, et al., Org. Chem. Front. 5 (2018) 998-1002. |
| [12] |
J.S. Sun, M. Liu, J. Zhang, L. Dong, J. Org. Chem. 83 (2018) 10555-10563. DOI:10.1021/acs.joc.8b01354 |
| [13] |
T. Yuan, C. Pi, C. You, X. Cui, et al., Chem. Commun. 55 (2019) 163-166. DOI:10.1039/C8CC08081J |
| [14] |
(a) L. Wang, D. Xiong, L. Jie, et al., Chin. Chem. Lett. 29 (2018) 907-910; (b) F. Xu, Y.J. Li, C. Huang, H.C. Xu, ACS Catal. 8 (2018) 3820-3824. |
| [15] |
V.K. Tiwari, N. Kamal, M. Kapur, Org. Lett. 17 (2015) 1766-1769. DOI:10.1021/acs.orglett.5b00535 |
| [16] |
T. Okada, K. Nobushige, T. Satoh, M. Miura, Org. Lett. 18 (2016) 1150-1153. DOI:10.1021/acs.orglett.6b00268 |
| [17] |
J. Jia, J. Shi, J. Zhou, X. Liu, et al., Chem. Commun. 51 (2015) 2925-2928. DOI:10.1039/C4CC09823D |
| [18] |
(a) L. Xu, T. Li, L. Wang, X. Cui, J. Org. Chem. 84 (2019) 560-567; (b) Z. Yang, L. Jie, Z. Yao, Z. Yang, X. Cui, Adv. Synth. Catal. 361 (2019) 214-218; (c) J. Xia, X. Yang, Y. Li, X. Li, Org. Lett. 19 (2017) 3243-3246; (d) C. Pi, X. Cui, X. Liu, M. Guo, et al., Org. Lett. 16 (2014) 5164-5167; (e) L. Li, W.W. Brennessel, W.D. Jones, Organometallics 28 (2009) 3492-3500. |