 2019, Vol. 30
2019, Vol. 30 


b Institute of Biology, Westlake Institute for Advanced Study, Hangzhou 310024, China
Venom toxins are a class of natural products that are isolated from a variety of vertebrates and invertebrates including scorpions, snakes, spiders, cone snails, sea anemones (Fig. 1) [1, 2].

|
Download:
|
| Fig. 1. Venomous animals that secret venom toxins for prey capture and defense. Copied with permission [1]. Copyright 2003, Springer Nature. | |
{kind=link}

|
Download:
|
| Fig. 2. Synthetic strategy of Ts3 variants. Copied with permission [20]. Copyright 2016, John Wiley and Sons. | |
{kind=link}
The natural reservoir for venom toxins is extremely large. It is estimated there are about 700 species of marine snails found in tropical and subtropical waters with each single species containing about100 peptideconotoxins[3, 4]. Evolutionarily, venom toxinsare produced in these animals for prey capture and defense [5]. They often target various physiologically important membrane proteins including ion channels, GPCRs, nicotinic acetylcholine receptors etc. [1, 5-8]. For that reason, they have been used to study the functional mechanism of these membrane proteins as well as to be developed as therapeutics. The applications of venom toxins have been reviewed by Lewis, Dutertre, Vetter, et al. in the past [9-12].
2. Chemical synthesis of venom toxinsTraditionally, venom toxins could only be obtained by milking venomous animals, which had limited the studies of these protein molecules. With the development and maturation of chemical protein synthesis [13], venom toxin studies have seen significant advance during the past two decades.
Venom toxins are ideal targets for chemical protein synthesis as has been discussed by Fang, Akondi, et al. [2, 3]. They are generally small, comprised of less than 100 amino acids, and rich in cysteines. Fortunately, these cysteines can be utilized in native chemical ligation to link different peptide segments together [14], facilitating synthesis of the linear polypeptides.
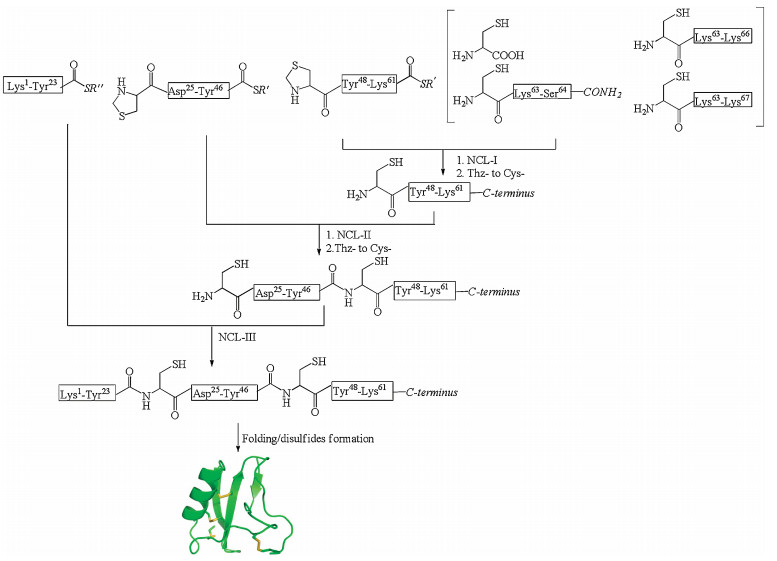
Synthesis of short venom toxins (< 30 residues) can be directly achieved using solid phase peptide synthesis [15] either through Boc chemistry [16] or Fmoc chemistry [17]. For longer venom toxin synthesis, convergent synthesis employing native chemical ligation is strongly preferred. Convergent synthesis gives higher yield for final products, as demonstrated by Kent, et al. for the one-pot convergent synthesis of Crambin [18]. Convergent synthesis is also more efficient for producing analogs since only the short modification-containing fragment must be resynthesized. We have adopted this strategy in recent years to synthesize ShK toxin, Ts1 toxin, and Ts3 toxin to explore their structure-activity relationships and to incorporate unnatural amino acids for sitespecific labeling of these toxins [19-21]. An example is shown in Fig. 2. In the synthesis of Ts3 toxin, the covalent structure of the Ts3 was uncertain since the toxin is post-translationally processed at the C-terminus [20, 22]. To resolve this confusion, we used native chemical ligation to prepare all the possible Ts3 variants. In the synthesis, we prepared the three different C-terminal peptides and were able to use all the other shared fragments to produce the four variants simultaneously within a short period of time [20]. Through activity test, we confirmed Ts3 has 64 residues with C-terminal amidation [20].
Similar strategy was taken for the synthesis and fluorescent dye labeling of Ts1 toxin. We first established three segments convergent synthesis, then explored different strategies to sitespecifically label Ts1 toxin with fluorescent dyes. Different sites were chosen and were changed to either cysteine or propargylglycine individually to explore labeling efficiencies. Because of the convergent synthetic strategy chosen, we only needed to produce short peptide segments for each labeling strategy. By using this approach, we were very rapidly able to determine the best way to label Ts1 [21]. While chemical synthesis of the primary sequence of venom toxins by various methodologies [14, 23-33] can range in difficulty, in many cases the more demanding bottleneck is the three-dimensional structure refolding. Venom toxins are rich in cysteines which form disulfide bonds to dictate their threedimensional structures. In the refolding process, we normally do not have control over the pairing of individual cysteines. Luckily, as Anfensen proposed and was later widely accepted, the structure of a protein molecule is encoded in its primary sequence [34]. If proper conditions can be provided, most wild type venom toxins can spontaneously fold into their correct structures with correct disulfides pairing.
Common initial refolding reaction could be performed by dissolving the polypeptide chain at dilute concentration (< 0.5 mmol/L) at alkaline pH (7.5-8.5). For example, air oxidation for ShK toxin [35, 36] refolding can be readily achieved in highyield with this method. Conotoxin MI can also be refolded in this way [37, 38].
The more popular and more broadly successful refolding conditions involve the addition of redox pairs, typically cysteine/cystine at 8 mmol/L/1 mmol/L or GSH/GSSG at 10 mmol/L/1 mmol/L. These redox pairs can help reshuffling the disulfide bonds thus facilitating their correct pairing. Often, 0.5-1 mol/L of GuHCl is added to increase the solubility of the folding intermediates. Many different venom toxins including BmBKTtx1 [39-41], kaliotoxin [42], GsMTx4 [43] have been refolded using the redox pairs.
However, for a number of other venom toxins, refolding can be more challenging. When we attempted the refolding of Ts1 toxin, polypeptide concentration had to be lowered down to 0.01 mg/mL and temperature needed to be dropped to 4 ℃ to achieve efficient refolding [21]. The folding of Ts3 toxin was even more problematic. In this case, 0.5 mol/L L-arginine was further added to decrease polypeptide aggregation [44]. We also found addition of 20% DMSO can greatly enhance Ts3 toxin folding yield.
One underlying principle for optimization of folding condition is to keep all folding intermediates in solution at all times. Adding GuHCl, L-arginine, DMSO, lowing temperature, polypeptide concentration are all different ways to increase polypeptide solubility, and to decrease aggregation that eventually may lead to precipitation. Other organic solvents which can increase the solubility of polypeptides may also be explored to achieve successful refolding [3].
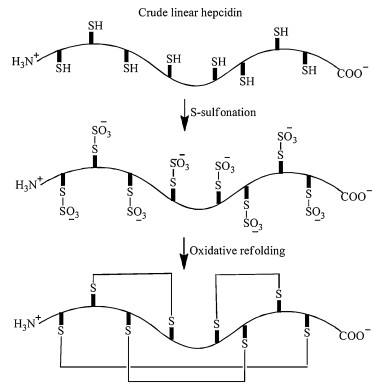
However, under some extreme situations, all these strategies can fail. One such example is the refolding of hepcidin (Fig. 3). Hepcidin primary sequence only has 25 amino acid residues, but with 8 cysteines forming 4 disulfide bonds. Air oxidation or DMSO oxidation at alkaline pH does not produce usable quantities of the folded product, since most of the polypeptides precipitate out of solution at basic pH [45]. Using redox pair GSH/GSSG, Miranda et al. could increase the folding yield to 6% which is not satisfactory. By employing the CLEAR-OX resin [46], they were able to refold hepcidin at pH 4.0 using crude linear polypeptide achieving 12% overall yield (Fig. 3) [45].

|
Download:
|
| Fig. 3. Different folding strategies of hepcidin. Reproduced with permission [45]. Copyright 2010, John Wiley and Sons. | |
{kind=link}
To tackle the refolding problem of hepcidin, Guo et al. took a different strategy (Fig. 4). They first performed S-sulfonation on crude hepcidin to introduce negatively charged sulfonate moieties on cysteines to increase hepcidin overall solubility [47]. Remarkably, when they then tried to fold sulfonated hepcidin under optimal folding conditions: 0.5 mol/L L-arginine, 1.0 mmol/L EDTA, pH 7.5, 30% acetonitrile, 0.5 mmol/L GSH, 0.5 mmol/L GSSG, 0.1 mg/mL linear polypeptide, 37 ℃, 4 h, the folding yield could reach up to 80% [47]. However, when we tried this strategy on the folding of Ts1 toxin, it did not have a dramatic effect on the overall folding yield. Thus, different strategies might need to be explored when difficult folding situation is encountered.

|
Download:
|
| Fig. 4. Folding hepcidin through S-sulfonation intermediates [47]. | |
{kind=link}
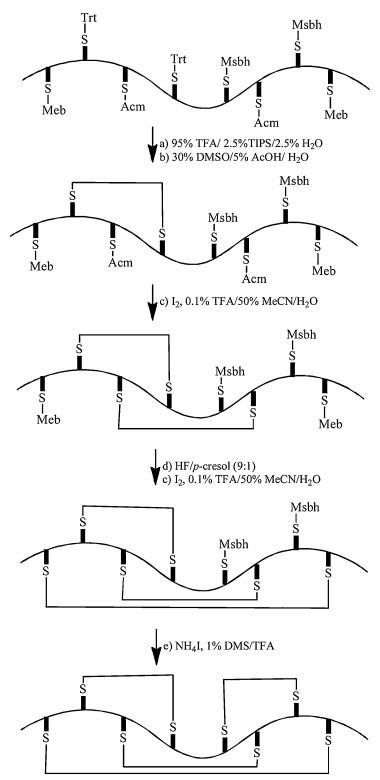
An alternative strategy to ensure correct disulfide bonds pairing is to perform regio-selective disulfide bonds formation using chemistry. Directed chemical synthesis of protein with three disulfides has been reported in a few cases [48, 49]. The directed synthesis of four disulfides has only recently been demonstrated for the synthesis of hepcidin [50]. Here, each pairing cysteine protecting groups is removed and the two unprotected cysteines are oxidized to form disulfide bond without touching other cysteines as shown in Fig. 5. This strategy demonstrates the elegance of modern chemical protein synthesis, but could only be achieved by skilled peptide chemists.

|
Download:
|
| Fig. 5. Disulfide directed chemical synthesis of hepcidin. Reproduced with permission [50]. Copyright 2014, John Wiley and Sons. | |
{kind=link}
3. Venom toxin structure determination
Following the successful synthesis of venom toxins, it is often desired to determine their three-dimensional structures to facilitate developing these protein molecules into therapeutic candidates or biophysical probes [1, 11].
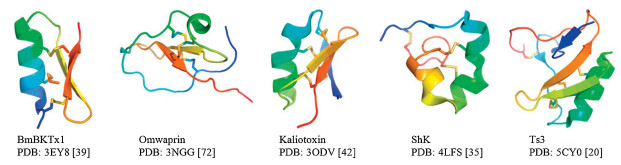
Traditionally, NMR and X-ray crystallography are the two methods of choice for venom toxin structure determination (Fig. 6) [51-59]. NMR is especially useful for structure determination of small venom toxins (e.g., conotoxins) since these molecules have short primary sequences; it is normally easy to assign all the amino acid peaks in the NMR spectrum [51]. However, when multiple disulfide bonds are present, assigning the correct connectivity of disulfide bonds could still be challenging [51].

|
Download:
|
| Fig. 6. Representative venom toxin structures solved by NMR (upper panel) and X-ray crystallography (lower panel). | |
{kind=link}
Many venom toxin structures have also been solved by using traditional X-ray crystallography, including alpha-scorpion toxin BMK M1 (pdb, 1SN1) [52], Ts1 toxin (pdb, 1B7D) [53], snake toxin Fasciculin 2 (pdb, 1FSC) [54], Bucandin (pdb, 1F94) [55] (Fig. 6). However, venom toxins generally have high percentage of charged amino acids, making them particularly soluble in water, obtaining high quality crystals has proven to be difficult in many situations because of the high solubility. For example, BmBKTx1 is a scorpion toxin that can block potassium channels [40, 41]. It was reported that crystallization screening of BmBKTx1 at 100 mg/mL using sparse-matrix, no precipitation was observed over many weeks at room temperature [40]. Reductive methylation had to be performed on lysine side chains and N-terminal amine to obtain crystals [40].
With the advance of chemical protein synthesis, preparing the mirror image forms of protein molecules has become readily accessible in recent years [13, 60]. This has provided an alternative choice for obtaining protein crystals since the two mirror image protein forms can be mixed together at 1:1 ratio to perform crystallization screening for faster crystal formation [60-62]. This concept of racemic protein crystallography was first proposed by Mackay [63] and was later demonstrated to be effective in small peptides [64-68]. In 2008, when Pentelute, et al. encountered the problem of obtaining sfAFP crystals and solving its structure, they adopted this racemic protein crystallography method and were able to produce crystals in ~50% of the screened conditions [69]. The structure was eventually solved by using quasi-racemic crystals. This work demonstrates racemicprotein crystallography can indeed significantly boost the success rate of obtaining high quality crystals from screening as has been predicated by Yeates [70].
The racemic-protein crystallography method has since been embraced for the structure determination of venom toxins (Fig. 7) [19, 20, 35, 39, 43, 71-73]. Facing the difficulty of producing BmBKTx1 crystals described above, Mandal, et al. employed chemical synthesis to readily prepare both L- and D-BmBKTx1. With the two mirror image protein forms in hands, they were able to obtain crystals overnight under multiple conditions [39]. When we were trying to grow Ts3 toxin crystals to determine its structure for choosing labeling sites, we faced the same difficulty. Screening hundreds of conditions failed to produce any crystal for L-Ts3 toxin. We then prepared the mirror image form D-Ts3 and performed racemic protein crystallization. Luckily, high quality crystals grew out from one condition among the hundreds of conditions screened [20]. These examples again demonstrate the power of racemic protein crystallography in determining venom toxin structures. Quasi-racemic protein crystallography provides a powerful tool for determining structures of protein analogs, where the same batch of wildtype D-protein can be re-used to form crystals with modified versions of the L-protein. As has been demonstrated in the sfAFP case, minimal change in the protein structure may not disturb the crystallization of the quasi-racemic protein mixture [69]. We used this method and determined a few ShK analog structures to explore the influence of side chain chirality change on ShK foldability, stability and three-dimensional structures [19]. We expect quasi-racemic crystallography to aid more structure-activity relationship studies of venom toxins in the future.

|
Download:
|
| Fig. 7. Representative venom toxin structures solved by racemic protein crystallography. | |
{kind=link}
Recent advance in Cryo-EM [74] technologies has made it possible to determine the structures of many difficult membrane proteins including eukaryotic voltage-gated calcium channel [75], voltage-gated sodium channel [76] and toxin-ion channel complexes [77-80]. In the future, we expect Cryo-EM will enable determination of more complex structures between venom protein toxins and their membrane protein targets. This will greatly deepen our understanding how venom toxins interact and modulate functions of membrane proteins to develop venom toxins as therapeutics as well as to better understand the target membrane protein function mechanisms.
4. ConclusionThe enormous resources of the venom toxins have been a gift from nature. Their diverse functions have been fascinating to scientists. With the advancement of chemical protein synthesis and structure determination, especially by racemic protein crystallography now and Cryo-EM in the future, these protein molecules should fulfill the role as future therapeutics and powerful biological probes.
| [1] |
R.J. Lewis, M.L. Garcia, Nat. Rev. Drug Discov. 2 (2003) 790-802. DOI:10.1038/nrd1197 |
| [2] |
G.M. Fang, X.X. Chen, Q.Q. Yang, et al., Chin. Chem. Lett. 29 (2018) 1033-1042. DOI:10.1016/j.cclet.2018.02.002 |
| [3] |
K.B. Akondi, M. Muttenthaler, S. Dutertre, et al., Chem. Rev. 114 (2014) 5815-5847. DOI:10.1021/cr400401e |
| [4] |
B.G. Livett, K.R. Gayler, Z. Khalil, Curr. Med. Chem. 11 (2004) 1715-1723. DOI:10.2174/0929867043364928 |
| [5] |
E.D. Brodie III, Curr. Biol. 19 (2009) R931-R935. DOI:10.1016/j.cub.2009.08.011 |
| [6] |
L. Luo, B. Li, S. Wang, et al., Proc. Natl. Acad. Sci. U. S. A. 115 (2018) 1646-1651. DOI:10.1073/pnas.1714760115 |
| [7] |
S. Luo, D. Zhangsun, P.J. Harvey, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) E4026-E4035. DOI:10.1073/pnas.1503617112 |
| [8] |
T. Yang, T. Xiao, Q. Sun, K. Wang, Acta Pharm. Sin. B 7 (2017) 611-622. DOI:10.1016/j.apsb.2017.09.001 |
| [9] |
I. Vetter, R.J. Lewis, Curr. Top. Med. Chem. 12 (2012) 1546-1552. DOI:10.2174/156802612802652457 |
| [10] |
C.I. Schroeder, D.J. Craik, Future Med. Chem. 4 (2012) 1243-1255. DOI:10.4155/fmc.12.70 |
| [11] |
S. Dutertre, R.J. Lewis, J. Biol. Chem. 285 (2010) 13315-13320. DOI:10.1074/jbc.R109.076596 |
| [12] |
B. Jouirou, A. Mosbah, V. Visan, et al., Biochem. J. 377 (2004) 37-49. DOI:10.1042/bj20030977 |
| [13] |
S.B. Kent, Chem. Soc. Rev. 38 (2009) 338-351. DOI:10.1039/B700141J |
| [14] |
P.E. Dawson, T.W. Muir, I. Clark-Lewis, S.B. Kent, Science 266 (1994) 776-779. DOI:10.1126/science.7973629 |
| [15] |
R.B. Merrifield, J. Am. Chem. Soc. 85 (1963) 2149-2154. DOI:10.1021/ja00897a025 |
| [16] |
M. Schnolzer, P. Alewood, A. Jones, D. Alewood, S.B. Kent, Int. J. Pept. Protein Res. 40 (1992) 180-193. |
| [17] |
R. Sheppard, J. Pept. Sci. 9 (2003) 545-552. |
| [18] |
D. Bang, S.B. Kent, Angew. Chem. Int. Ed. Engl. 43 (2004) 2534-2538. |
| [19] |
B. Dang, R. Shen, T. Kubota, et al., Angew. Chem. Int. Ed. 56 (2017) 3324-3328. DOI:10.1002/anie.201612398 |
| [20] |
B. Dang, T. Kubota, K. Mandal, et al., Angew. Chem. Int. Ed. 55 (2016) 8639-8642. DOI:10.1002/anie.201603420 |
| [21] |
B. Dang, T. Kubota, A.M. Correa, F. Bezanilla, S.B. Kent, Angew. Chem. Int. Ed. 53 (2014) 8970-8974. DOI:10.1002/anie.201404438 |
| [22] |
M.F. Martin-Eauclaire, B. Ceard, A.M. Ribeiro, et al., FEBSLett. 342 (1994) 181-184. DOI:10.1016/0014-5793(94)80496-6 |
| [23] |
G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. DOI:10.1002/anie.201100996 |
| [24] |
J.B. Blanco-Canosa, P.E. Dawson, Angew. Chem. Int. Ed. 47 (2008) 6851-6855. |
| [25] |
J.B. Blanco-Canosa, B. Nardone, F. Albericio, P.E. Dawson, J. Am. Chem. Soc. 137 (2015) 7197-7209. DOI:10.1021/jacs.5b03504 |
| [26] |
V.R. Pattabiraman, A.O. Ogunkoya, J.W. Bode, Angew. Chem. Int. Ed. 51 (2012) 5114-5118. DOI:10.1002/anie.201200907 |
| [27] |
Y. Zhang, C. Xu, H.Y. Lam, C.L. Lee, X. Li, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 6657-6662. DOI:10.1073/pnas.1221012110 |
| [28] |
N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain, O. Melnyk, Org. Lett. 12 (2010) 5238-5241. DOI:10.1021/ol102273u |
| [29] |
A. Brust, C.I. Schroeder, P.F. Alewood, ChemMedChem 9 (2014) 1038-1046. DOI:10.1002/cmdc.201300524 |
| [30] |
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. DOI:10.1002/anie.201203843 |
| [31] |
J.S. Zheng, S. Tang, Y.K. Qi, Z.P. Wang, L. Liu, Nat. Protoc. 8 (2013) 2483-2495. DOI:10.1038/nprot.2013.152 |
| [32] |
M. Pan, Y. He, M. Wen, et al., Chem. Commun. 50 (2014) 5837-5839. DOI:10.1039/C4CC00779D |
| [33] |
H. Lan, K. Wu, Y. Zheng, et al., J. Pept. Sci. 22 (2016) 320-326. DOI:10.1002/psc.2868 |
| [34] |
M. Sela, F.H. White Jr., C.B. Anfinsen, Science 125 (1957) 691-692. DOI:10.1126/science.125.3250.691 |
| [35] |
B. Dang, T. Kubota, K. Mandal, F. Bezanilla, S.B. Kent, J. Am. Chem. Soc. 135 (2013) 11911-11919. DOI:10.1021/ja4046795 |
| [36] |
M.W. Pennington, M.E. Byrnes, I. Zaydenberg, et al., Int. J. Pept. Protein Res. 46 (1995) 354-358. |
| [37] |
G. Bulaj, B.M. Olivera, Antioxid. Redox Signal. 10 (2008) 141-155. DOI:10.1089/ars.2007.1856 |
| [38] |
W.R. Gray, J.E. Rivier, R. Galyean, L.J. Cruz, B.M. Olivera, J. Biol. Chem. 258 (1983) 12247-12251. |
| [39] |
K. Mandal, B.L. Pentelute, V. Tereshko, A.A. Kossiakoff, S.B. Kent, J. Am. Chem. Soc. 131 (2009) 1362-1363. DOI:10.1021/ja8077973 |
| [40] |
A. Szyk, W. Lu, C. Xu, J. Lubkowski, J. Struct. Biol. 145 (2004) 289-294. DOI:10.1016/j.jsb.2003.11.012 |
| [41] |
Z. Cai, C. Xu, Y. Xu, et al., Biochemistry 43 (2004) 3764-3771. DOI:10.1021/bi035412+ |
| [42] |
B.L. Pentelute, K. Mandal, Z.P. Gates, et al., Chem. Commun. 46 (2010) 8174-8176. DOI:10.1039/c0cc03148h |
| [43] |
Q. Qu, S. Gao, Y.M. Li, J. Pept. Sci. 24 (2018) e3112. |
| [44] |
C. Lange, R. Rudolph, Curr. Pharm. Biotechnol. 10 (2009) 408-414. DOI:10.2174/138920109788488851 |
| [45] |
J. Zhang, S. Diamond, T. Arvedson, B.J. Sasu, L.P. Miranda, Biopolymers 94 (2010) 257-264. DOI:10.1002/bip.21383 |
| [46] |
K. Darlak, D. Wiegandt Long, A. Czerwinski, et al., J. Pept. Res. 63 (2004) 303-312. DOI:10.1111/j.1399-3011.2004.00153.x |
| [47] |
X. Luo, Q. Jiang, G. Song, et al., FEBS J. 279 (2012) 3166-3175. DOI:10.1111/j.1742-4658.2012.08695.x |
| [48] |
H. Gali, G.L. Sieckman, T.J. Hoffman, et al., Bioconjug. Chem. 13 (2002) 224-231. DOI:10.1021/bc010062u |
| [49] |
C. Kellenberger, H. Hietter, B. Luu, Pept. Res. 8 (1995) 321-327. |
| [50] |
Z. Dekan, M. Mobli, M.W. Pennington, et al., Angew. Chem. Int. Ed. 53 (2014) 2931-2934. DOI:10.1002/anie.201310103 |
| [51] |
A. Walewska, J.J. Skalicky, D.R. Davis, et al., J. Am. Chem. Soc. 130 (2008) 14280-14286. DOI:10.1021/ja804303p |
| [52] |
X.L. He, H.M. Li, Z.H. Zeng, et al., J. Mol. Biol. 292 (1999) 125-135. DOI:10.1006/jmbi.1999.3036 |
| [53] |
I. Polikarpov, M.S. Junior, S. Marangoni, et al., J. Mol. Biol. 290 (1999) 175-184. DOI:10.1006/jmbi.1999.2868 |
| [54] |
M.H. le Du, D. Housset, et al., Acta Crystallogr.D-Biol.Crystallogr. 52 (1996) 87-92. DOI:10.1107/S0907444995007517 |
| [55] |
P. Kuhn, A.M. Deacon, S. Comoso, et al., Acta Crystallogr. D-Biol. Crystallogr. 56 (2000) 1401-1407. DOI:10.1107/S0907444900011501 |
| [56] |
I.A. Sharpe, J. Gehrmann, M.L. Loughnan, et al., Nat. Neurosci. 4 (2001) 902-907. DOI:10.1038/nn0901-902 |
| [57] |
A. Nicke, M.L. Loughnan, E.L. Millard, et al., J. Biol. Chem. 278 (2003) 3137-3144. DOI:10.1074/jbc.M210280200 |
| [58] |
A. Prochnicka-Chalufour, G. Corzo, H. Satake, et al., Biochemistry 45 (2006) 1795-1804. DOI:10.1021/bi0519248 |
| [59] |
N.S. Bende, S. Dziemborowicz, M. Mobli, et al., Nat. Commun. 5 (2014) 4350. DOI:10.1038/ncomms5350 |
| [60] |
T.O. Yeates, S.B. Kent, Annu. Rev. Biophys. 41 (2012) 41-61. DOI:10.1146/annurev-biophys-050511-102333 |
| [61] |
S.B. Kent, Curr. Opin. Chem. Biol. 46 (2018) 1-9. |
| [62] |
B. Yan, L. Ye, W. Xu, L. Liu, Bioorg. Med. Chem. 25 (2017) 4953-4965. DOI:10.1016/j.bmc.2017.05.020 |
| [63] |
A.L. Mackay, Nature 342 (1989) 133-133. |
| [64] |
W.R. Patterson, D.H. Anderson, W.F. DeGrado, D. Cascio, D. Eisenberg, Protein Sci. 8 (1999) 1410-1422. |
| [65] |
L.W. Hung, M. Kohmura, Y. Ariyoshi, S.H. Kim, J. Mol. Biol. 285 (1999) 311-321. DOI:10.1006/jmbi.1998.2308 |
| [66] |
C. Toniolo, C. Peggion, M. Crisma, et al., Nat. Struct. Biol. 1 (1994) 908-914. DOI:10.1038/nsb1294-908 |
| [67] |
M. Doi, A. Ishibe, H. Shinozaki, et al., Int. J. Pept. Protein Res. 43 (1994) 325-331. |
| [68] |
L.E. Zawadzke, J.M. Berg, Proteins-Structure Function and Genetics 16 (1993) 301-305. |
| [69] |
B.L. Pentelute, Z.P. Gates, V. Tereshko, et al., J. Am. Chem. Soc. 130 (2008) 9695-9701. DOI:10.1021/ja8013538 |
| [70] |
S.W. Wukovitz, T.O. Yeates, Nat. Struct. Biol. 2 (1995) 1062-1067. DOI:10.1038/nsb1295-1062 |
| [71] |
C.K. Wang, G.J. King, S.E. Northfield, P.G. Ojeda, D.J. Craik, Angew. Chem. Int. Ed. 53 (2014) 11236-11241. DOI:10.1002/anie.201406563 |
| [72] |
J.R. Banigan, K. Mandal, M.R. Sawaya, et al., Protein Sci. 19 (2010) 1840-1849. DOI:10.1002/pro.468 |
| [73] |
K. Mandal, B.L. Pentelute, V. Tereshko, et al., Protein Sci. 18 (2009) 1146-1154. DOI:10.1002/pro.127 |
| [74] |
Y. Cheng, N. Grigorieff, P.A. Penczek, T. Walz, Cell 161 (2015) 438-449. DOI:10.1016/j.cell.2015.03.050 |
| [75] |
J. Wu, Z. Yan, Z. Li, et al., Nature 537 (2016) 191-196. DOI:10.1038/nature19321 |
| [76] |
H. Shen, Q. Zhou, X. Pan, et al., Science 355 (2017) eaal4326. DOI:10.1126/science.aal4326 |
| [77] |
H. Shen, Z. Li, Y. Jiang, et al., Science 362 (2018) eaau2596. DOI:10.1126/science.aau2596 |
| [78] |
D. Sun, Y. Yu, X. Xue, et al., Cell Discov. 4 (2018) 27. |
| [79] |
X. Pan, Z. Li, X. Huang, et al., Science 363 (2019) 1309-1313. DOI:10.1126/science.aaw2999 |
| [80] |
H. Shen, D. Liu, K. Wu, J. Lei, N. Yan, Science 363 (2019) 1303-1308. DOI:10.1126/science.aaw2493 |