 2019, Vol. 30
2019, Vol. 30 


b Henan Nonferrous Metals Geological Exploration Institute, Zhengzhou 450052, China

Xiao-Lan Chen received her PhD degree under the supervision of Prof. Yu-Fen Zhao at Zhengzhou University. She had worked as a researcher in University of Reading and visiting scholar at University of Southern California before joining Zhengzhou University in 2004. Her research interest is developing highly efficient methodologies for the synthesis of P-, S-, F-containing organic compounds;

Bing Yu received Bachelor's degree (2009) from Central China Normal University, China, and obtained his pH.D. (2014) under the supervision of Prof. Liang-Nian He at Nankai University, China. He then moved to Beijing Institute of Technology (2014–2016), Seoul National University (2015–2016) and Tokyo Institute of Technology (2016–2017) as a postdoctoral researcher. He joined Zhengzhou University in 2017. His research interest is developing novel approaches for green and efficient organic synthesis.
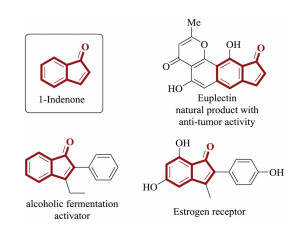
1-Indenones are privileged structural scaffolds that can be widely found in natural products and a large variety of biologically active molecules, such as alcoholic fermentation activators and estrogen binding receptors (Scheme 1) [1, 2]. Traditional methods for the construction of 1-indenones heavily relied on intramolecular Friedel-Crafts acylation [3], Heck-Larock cyclization reactions [4, 5], and Grignard reagent-initiated reactions [6, 7]. However, those classical methods suffer from harsh reaction conditions and practical inconvenience. Therefore, simple and efficient methods for the construction of 1-indenones are highly desired from a synthetic practicality viewpoint.

|
Download:
|
| Scheme 1. Representative examples of 1-indenones. | |
{kind=link}
In past decades, the development of synthetic methods for the preparation of 1-indenones scaffold has drawn much attention [8-20]. For example, the conventional method for the construction of 1-indenones generally relied on the design of functionalized alkynes and aromatic reaction partners (i.e., iodobenzaldehydes, ortho-bifunctionalized arenes) under the catalysis of precious metal catalysts (Rh, Pd, etc.). However, when unsymmetrical alkynes were applied, the poor regioselectivity may limit their synthetic application (Scheme 2a).

|
Download:
|
| Scheme 2. Representative synthetic strategies for the construcion of 1-indenones. | |
{kind=link}
Recently, radical chemistry has emerged as a powerful and versatile tool for organic synthesis [21-23]. Particularly, radical cascade reactions have been the subject of intense research because of their wide applications in the rapid construction of complicated carbocyclic and heterocyclic frameworks [24-30]. For instance, the addition of radicals to unsaturated C≡C triple bonds is well-known process, which will furnish the alkene radical species followed by cascade cyclization. Consequently, the construction of 1-indenones could be triggered by the in situ generated radicals and alkyne-containing substrates in one-pot cascade manner (Scheme 2b). This type of radical cyclization reactions generally initiated by the radical precursors and appropriate initiators, avoiding the use of noble metal catalysis [31-35].
From the standpoint of practical and green chemistry, the development of operationally simple, highly selective procedures for organic synthesis starting from easily available and inexpensive reagents is extremely meaningful and attractive [36-43]. In the recent years, construction of valuable and versatile 1-indenones via radical cascade reaction has attracted significant scientific interest from both medicinal and chemical scientists. The remarkable advantages of radical cascade reactions include atom/step economy as well as simple operation and cost reduction. This is a vibrant and fast-expanding field, which has not been well documented so far. Therefore, in this review, we will focus on recent advances in the synthesis of functionalized 1-indenones via radical cascade reactions.
2. 1, 3-Diarylpropynones as substrate1, 3-Diarylpropynones 1 are an important class of building blocks in organic synthesis, which can be conveniently prepared through Sonogashira coupling by the reactions of acyl chlorides and terminal alkynes [44-46]. Recently, radical cascade cyclization reactions of 1, 3-diarylpropynones with different radical precursor, such as phosphine oxides, alkanes, sulfinic acids, sulfonyl hydrazides, cyclic ethers etc., have been demonstrated to be efficient methods for the synthesis of functionalized 1-indenones.
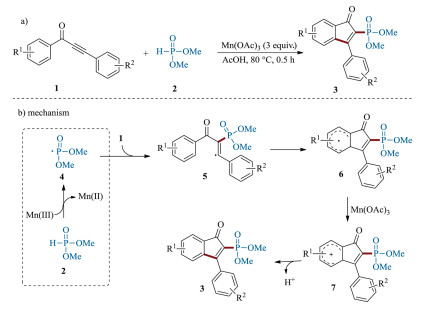
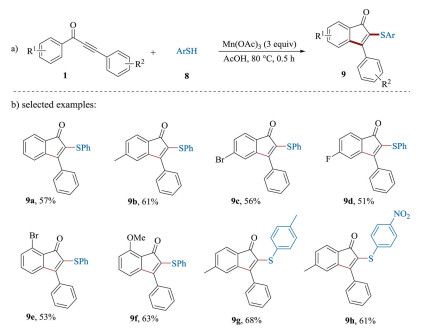
Organophosphorus compounds have been widely found in a large variety of pharmaceutically active molecules, agrochemicals and organic materials [47-52]. The phosphorus substituents can not only modify biological responses or medicinal properties, but also can act as ligands of transition metals [53-58]. Therefore, the development of methods for the incorporation of phosphoryl substituents into organic molecular is highly desired [59-61]. In this regard, in 2011 the Zou's group first reported an efficient Mn (Ⅲ)-mediated procedure for the construction of 2-phosphonylated 1-indenones 3 by reacting 1, 3-diarylpropynones 1 with dimethyl phosphonate 2 (Scheme 3a) [62, 63]. A plausible mechanism was proposed as shown in Scheme 3b. Initially, in the presence of Mn(OAc)3, dimethyl phosphonate 2 underwent a single electron transfer (SET) to generate the corresponding phosphoryl radical 4. Then, the addition of radical 4 to the C≡C triple bond of 1, 3-diarylpropynones 1 delivered the intermediate 5. Subsequently, an intramolecular cyclization of 5 gave intermediate 6, which was further oxidized into the carbocation 7 by Mn(Ⅲ) via a SET process. Finally, a rapid deprotonation of carbocation 7 regenerated the aromatic ring to yield the product 3. Notable, this elegant protocol is also suitable for the preparation of 2-thiolated 1-indenones 9 by using aryl thiols 8 as radical precursor through the similar reaction pathway (Scheme 4) [63]. Under the optimized reaction conditions, a large variety of 2-thiolated 1-indenones were prepared in moderate yields.

|
Download:
|
| Scheme 3. The construction of 2-phosphonylated 1-indenones. | |
{kind=link}

|
Download:
|
| Scheme 4. The preparation of 2-thiolated 1-indenones. | |
{kind=link}
Since this pioneering study, reactivity of 1, 3-diarylpropynones with various radical precursors has been intensively investigated. In 2016, Yu and co-workers developed a metal-free system with benzoyl peroxide (BPO) as the sole oxidant to synthesize a series of 2-alkylated 1-indenones 11 from 1, 3-diarylpropynones by using alkanes 10 as radical precursors (Scheme 5a) [64]. Under the optimized conditions, the corresponding products 11 were obtained in moderate to good yields with high regio- and stereo-selectivity. The representative examples are summarized in Scheme 5b. Cyclopentane, cyclohexane, cycloheptane and cyclooctane were all worked well (11a-d). Unfortunately, when acyclic alkanes, such as pentane and 2-methylbutane, were employed as substrates, the selectivity of the reaction is not good (11e-f).

|
Download:
|
| Scheme 5. The preparation of 2-alkylated 1-indenones. | |
{kind=link}
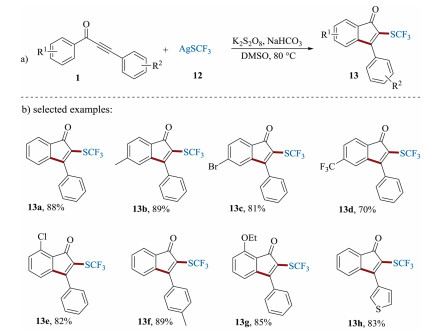
At the same year, Zhang et al. reported the silver-mediated trifluoromethylthiolation of 1, 3-diarylpropynones by employing AgSCF3 12 as a radical precursor (Scheme 6) [65]. This method proceeds in DMSO at 80 ℃ for 12 h to access 24 examples of 2-(trifluoromethylthio)-1-indenones 13 with moderate to excellent yields. Due to the importance of fluorine-containing 1-indenones, this protocol for introducing trifluoromethylthiol group into the 1-indenones displays potential for further application in synthetic and medicinal chemistry.

|
Download:
|
| Scheme 6. Synthesis of 2-(trifluoromethylthio)-1-indenones. | |
{kind=link}
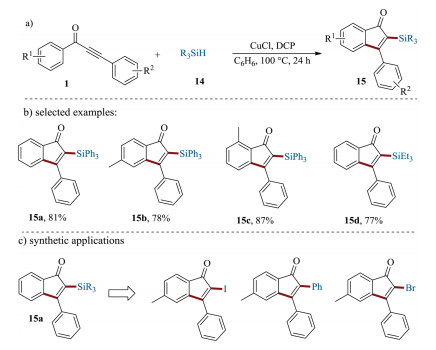
Recently, a copper-catalyzed protocol to access silyl-functionalized 1-indenones with commercially available silanes as radical precursors was disclosed by Zhu et al. (Scheme 7a) [66]. This method represents an efficient route to access a wide range of silylated 1-indenones in moderate to high yields with broad scope, excellent selectivity as well as gram scale-up ability (Scheme 7b). Notably, these silylated 1-indenones can act as versatile building blocks for valuable organic transformations (Scheme 7c). The KIE studies revealed that generation of silyl radical by the cleavage of Si-H bond would be the rate-determining step in this transformation.

|
Download:
|
| Scheme 7. The construction of silyl-functionalized 1-indenones. | |
{kind=link}
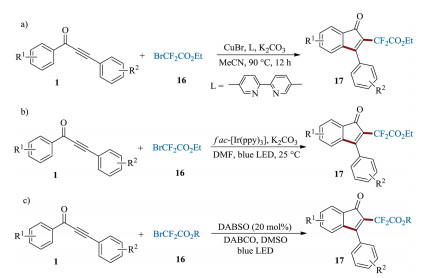
Development of efficient methodologies for the incorporation of the fluorine-containing functional groups into heterocyclic skeleton is considerably attractive due to their important role in altering physical and physiological properties [67]. In 2017, Zhang et al. reported a copper-catalyzed cascade difluoromethylation of 1, 3-diarylpropynones, allowing the direct introducing of difluoromethylene groups ('CF2') and construction of an 1-indenones scaffold in one-pot manner (Scheme 8a) [68]. Alternatively, a visible-light photoredox catalysis system has been developed recently as a green and efficient tool for the construction of valuable difluoromethylene moiety containing 1-indenones skeletons. For example, Rastogi et al. reported the synthesis of 2-difluoroacetylated 1-indenones 17 via a visible-light-induced difluoromethylation/cyclization of 1, 3-diarylpropynones catalyzed by Ir photocatalyst (Scheme 8b) [69]. Almost at the same time, Wu's group developed a DABSO-catalyzed photoinduced reaction to access 2-difluoroacetylated 1-indenones (Scheme 8c) [70]. Compared to Zhang's method, this metal-free photoredox system provides a more sustainable and convenient alternative for the difluoromethylation of 1-indenone scaffolds.

|
Download:
|
| Scheme 8. The preparation of 2-difluoroacetylated 1-indenones. | |
{kind=link}
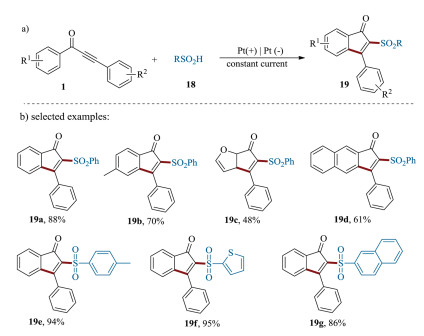
In addition to above radical precursors, sulfonyl radical have found to be an efficient reaction partner, which could be generated from sulfinic acids, sulfonyl hydrazides, sodium metabisulfite as precursors [71, 72]. Very recently, the group of Lei disclosed a novel and facile electrooxidative synthetic methodology to access a large variety of 2-sulfonated 1-indenones 19, by reacting various 1, 3-diarylpropynones 1 and sulfinic acids 18 (Scheme 9) [73]. Various functional groups could be tolerated under this mild reaction conditions, showing the potential for convenient and efficient preparation of sulfonyl containing 1-indenones.

|
Download:
|
| Scheme 9. Electrochemical synthesis of 2-sulfonated 1-indenones. | |
{kind=link}
3. 2-Alkynylbenzonitriles as substrate
Radical cascade cyclization of 2-alkynylbenzonitriles 20 with different radical precursors is another attractive strategy for the construction of 1-indenones. In 2016, Tu and Jiang successfully reported a novel silver-mediated radical cyclization of 2-alkynylbenzonitriles 20 with phosphine oxides towards 3-phosphinylated 1-indenones 22 (Scheme 10a) [74]. It is noteworthy that the reaction has obvious advantages such as broad substrate scope, mild reaction conditions and high functional group tolerances. As can be found in Scheme 10b, a reasonable mechanism was suggested. The phosphine oxide 21 can produce phosphoruscentered radical 23 by reacting with Ag(Ⅰ) through a SET process. After that, the regioselective addition of radical 23 to the C≡C triple bond of 2-alkynylbenzonitriles 20 formed the alkenyl radical 24, which underwent an intramolecular cyclization, rendering the intermediate 25. Radical 25 subsequently abstracted a hydrogen atom to yield imine 26, which then was converted to the target product 22 via hydrolysis in the presence of H2O.

|
Download:
|
| Scheme 10. Construction of 3-phosphinylated 1-indenones. | |
{kind=link}
In following contributions, the same group demonstrated a novel approach to access 3-alkylated 1-indenones 29 via radical cascade cyclization of 2-alkynylbenzonitriles with cyclic ethers (Scheme 11a) [75]. Under the optimized reaction conditions, a large variety of 3-alkylated 1-indenones 29 were prepared with good to excellent yields. Moreover, 1, 3-dioxolane, tetrahydrofuran and 1, 4-dioxane were well tolerated (Scheme 11b). Remarkably, cyclic ethers not only act as the C-centered radical precursors but also play the role of reaction media. Importantly, this reaction system employed the excess amount of 4-ClC6H4SO2Na as a unique additive, which was generally regarded as a precusor of sulfonyl radical. However, in this protocol the in situ generated sulfonyl radical can be tolerated, and played a significant role to promote the formation of C-centered radicals intrigging the radical cascade reactions.

|
Download:
|
| Scheme 11. Synthesis of 3-alkylated 1-indenones. | |
{kind=link}
As mentioned above, sulfone moieties represent a privileged structure with unique chemical properties and biological activities, which are widely found in organic synthesis, agrochemicals, pharmaceutical industry and materials science [76-80]. In the recent years, the introduction of sulfonyl groups into aromatic compounds has attracted more and more interests. In this context, almost at the same time, Tu's group, Liang's group and our group independently reported the synthesis of 3-sulfonyl 1-indenones 31 via sulfonyl radical initiated cascade cyclization of 2-alkynylbenzonitriles. In the study reported by Tu et al., Cu(OTf)2 has been applied as the catalyst and sulfonyl hydrazides 30 were the sulfonyl radical precursors (Scheme 12a) [81]. A plausible mechanism was proposed based upon the experimental outcomes as shown in Scheme 12b. Sulfonyl radicals 34 can be produced from sulfonyl hydrazides 30 in the presence of Cu(Ⅱ) and TBHP. Then, with the assistance of Cu(Ⅱ), the addition of radical 34 to the C≡C triple bond of 20 formed the alkenyl radical 35. After an intramolecular cyclization and hydrogen atom abstraction, the intermediate 37 was generated, followed by hydrolysis affording the desired product 31.

|
Download:
|
| Scheme 12. Preparation of 3-sulfonated 1-indenones. | |
{kind=link}
Liang's group developed a sulfonyl radical initiated cascade cyclization for the synthesis of 3-sulfonated 1-indenones by employing sodium sulfinates 38 as sulfonyl radical sources in the presence of Na2S2O8 as oxidant under transition metal-free conditions (Scheme 13) [82]. The sulfonyl radicals were generated from 38 under the oxidation of Na2S2O8. A large number of 3-sulfonated 1-indenones were prepared in moderate to good yields. More importantly, when sodium trifluoromethanesulfinate was employed as radical sources, trifluoromethylated 1-indenone 31 was obtained in 35% yield.

|
Download:
|
| Scheme 13. Synthesis of 3-sulfonated 1-indenones. | |
{kind=link}
For the reaction of 2-alkynylbenzonitriles 20 and sulfonyl hydrazides 30 to synthesize 3-sulfonyl 1-indenones 31, a more practical method was developed independently by our group. In this method, CuI was employed as catalyst, playing extremely important role in this transformation. Different from the mechanism proposed by Tu's group, the key intermediate 39 was proposed to be obtained by the coupling of radical 36 and Cu(OH)I. Then, intermediate 39 abstracts a H-atom from TBHP to generate imine intermediate 37 (Scheme 14). All of these pathways were systematically studied by DFT calculations. More importantly, these 3-sulfonyl 1-indenones were serendipitously found to own typical aggregation-induced emission (AIE) properties in our research process, showing orange to red emission with a large Stokes shift. In addition, this novel AIEgens exhibit excellent biocompatibility, which can be successfully used in live cell imaging [83].

|
Download:
|
| Scheme 14. Preparation of 3-sulfonated 1-indenones. | |
{kind=link}
4. Arylpropynols as substrate
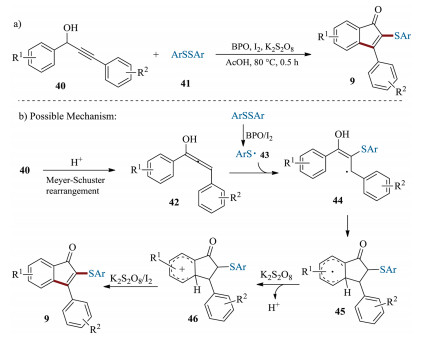
In 2016, Zhang et al. developed an iodine-mediated radical cascade cyclization for the synthesis of 2-thiolated indenones 9 (Scheme 15) [84]. The reaction was applied to a large variety of arylpropynols and disulfides, providing corresponding 2-thiolated 1-indenones 9 in moderate to good yields. Compared with the previous protocol [63], odorless disulfides, rather than the odorous thiol compounds, were employed as radical precursors. A plausible mechanism was proposed for this transformation as shown in Scheme 15b. Initially, the propargyl alcohol 40 is converted to an active allenol intermediate 42 in the presence of acid via Meyer-Schuster rearrangement. Then, sulfenyl radical 43, which was generated from disulfide 41, selectively attacks the C=C bond of intermediate 42 to afford radical 44. Then, 44 subsequently undergoes keto-enol tautomerization and the radical cyclization to give radical 44, which can be oxidized to the corresponding product 9.

|
Download:
|
| Scheme 15. The preparation of 2-thiolated 1-indenones. | |
{kind=link}
5. 1, 5-Enynes as substrate
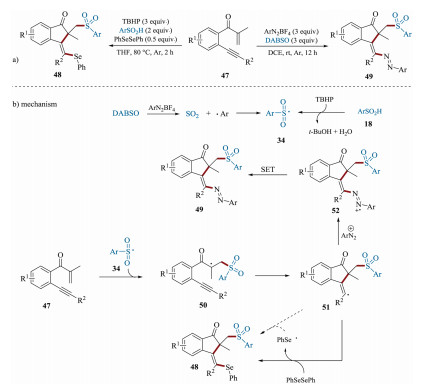
In 2018, Tu's group disclosed a straightforward protocol for the preparation of sulfonated 1-indenones [85] (Scheme 16a). In this synthetic methodology, lots of sulfonated 1-indenones were regioselectively obtained in moderate to good yields. More importantly, under oxidant-free conditions, azosulfonylation of 1, 5-enynes 47 was successfully realized, showing high functional group tolerance. As shown in Scheme 16b, the mechanism for the azosulfonylation of 1, 5-enynes showed that arylsulfonyl radical 34 can be generated via the reactions of DABSO and aryldiazonium salts. Then, the addition of radical 34 to the C=C bonds of 47 forms intermediate 50. After an intramolecular cyclization, vinyl radical 51 was produced, which subsequently trapped by aryldiazonium cations to access radical cation 52. The intermediate 52 can further undergo a SET process to give the corresponding product 49. For the formation of 48, similar mechanism involving radical addition and cyclization as well as radical coupling was proposed. This strategy opens a brand-new area for the construction of functionalized 1-indenones with advantages of step/atom economy, experimental simplicity and easy workup.

|
Download:
|
| Scheme 16. The preparation of sulfonated 1-indenones. | |
{kind=link}
It is well known that visible-light photoredox catalysis has been considered as more attractive synthetic tool, due to the merits of safety, green and sustainability. Following the increased interest in the construction of 1-indenones, the same group developed a novel and efficient visible-light photocatalytic synthetic methodology, by which a number of syn-fluoren-9-ones were prepared via reactions of 1, 5-enynes 47 and α-bromomalonates 53 [86] (Scheme 17a). As seen in Scheme 17b, under visible-light irradiation, fac-[IrⅢ(ppy)3] is excited to form fac*[IrⅢ(ppy)3], which reduces 53 to form radical 55 via SET process. After radical addition and cyclization, vinyl radicals 57 was produced, which undergoes 1, 6-H atom transfer (HAT), followed by 6-endo-trig cyclization to give radical intermediates 59. Radical intermediates 59 were further oxidized into the carbocations 60 by fac-[IrⅣ(ppy)3], along with the regeneration of fac-[IrⅢ(ppy)3] for the next cycle. Finally, the final products 54 were formed by deprotonation of carbocations 60.

|
Download:
|
| Scheme 17. The preparation of syn-fluoren-9-ones. | |
{kind=link}
6. 2-Alkynylated bromocinnamates as substrate
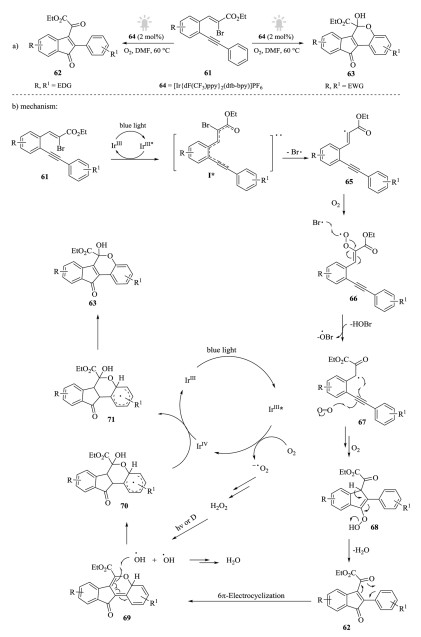
In 2017, Reiser et al. developed visible-light-mediated photocascade reaction for the preparation of 1-indenones and dihydroindeno[1, 2-c]chromenes in one-pot manner [87] (Scheme 18a). Interestingly, 2-alkynylated bromocinnamates 61 with electrondonating groups (EDG) were transformed into 1-indenones 62, while those with electron-withdrawing groups (EWG) were converted to dihydroindeno[1, 2-c]chromenes 63. Control experiments demonstrated that the oxygen atoms were derived from O2 by employing 18O labeled oxygen. A mechanism for this transformation was proposed in Scheme 18b. The energy transfer between the excited *IrⅢ and 61 generated activated vinyl bromide species I*, which lose Br* providing vinyl radical 65. This radical was confirmed by a trapping experiment with TEMPO and EPR. Then, 65 was capable of trapping O2 to give intermediate 66, which subsequently decompose into intermediate 67. After 5-endo-dig cyclization followed by the trapping of O2, intermediate 68 was formed, which yield the products 62 by elimination of water. Moreover, products 62 bearing electron-withdrawing substituents undergo 6π-electrocyclization to generate 63.

|
Download:
|
| Scheme 18. Synthesis of indenones and dihydroindeno[1, 2-c]chromenes. | |
{kind=link}
7. Conclusions and outlook
In summary, significant progress has been made in radical cascade reaction for the construction of 1-indenones over the past few years. By employing 1, 3-diarylpropynones, 2-alkynylbenzonitriles, arylpropynols, 1, 5-enynes and 2-alkynylated bromocinnamates as substrates for the radical cascade reactions, a large number of 1-indenones derivatives including 2-phosphonyl, 2-thiol, 2-alkyl, 2-trifluoromethylthiol, 2-silyl, 2-difluoroacetyl, 3-phosphonyl, 2-sulfonyl, 3-alkyl, 3-sulfonyl functionalized 1-indenones were efficiently constructed in efficient one-pot manner. This review has summarized the recent research carried out in this area. It can be seen that synthetic work has been mainly focused on P-, Si-, C- and S-centered radicals involved cascade reactions for the synthesis of functionalized 1-indenones, whereas the synthetic potential of O-, N-, B-centered radicals have never been explored in this context. Further expansion of this meaningful methodology for the construct more structure diverse 1-indenones will be highly expected. In addition, oxidative conditions for the generation of radicals are usually suffering from stoichiometric strong oxidants as well as high reaction temperatures, which greatly limiting their further application. Considering the high complexity of natural products and pharmaceuticals, it is still highly enthusiastically pursued and expected to develop efficient synthetic strategies for the total synthesis of those compounds under mild and greener reaction conditions, such as photoredox catalysis and electrocatalysis. We believe that the rapid development of radical chemistry will open new avenues for the construction of more structure diverse 1-indenones in the future.
AcknowledgmentsWe acknowledge the financial support from the National Natural Science Foundation of China (No. 21501010), Key Research Projects of Universities in Henan Province (No. 19A350011), 2017 Science and Technology Innovation Team in Henan Province (No. 22120001), Major Scientific and Technological Projects in Henan Province (No. 181100310500).
| [1] |
A.V. Vasilyev, S. Walspurger, P. Pale, J. Sommer, Tetrahedron Lett. 45 (2004) 3379-3381. DOI:10.1016/j.tetlet.2004.03.026 |
| [2] |
Y. Sheng, Y. You, Z. Cao, L. Liu, H.C. Wu, Analyst 143 (2018) 2411-2415. DOI:10.1039/C8AN00580J |
| [3] |
M.B. Floyd, G.R. Allen, J. Org. Chem. 35 (1970) 2647-2653. DOI:10.1021/jo00833a036 |
| [4] |
W. Tao, L.J. Silverberg, A.L. Rheingold, R.F. Heck, Organometallics 8 (1989) 2550-2559. DOI:10.1021/om00113a006 |
| [5] |
R.C. Larock, Q. Tian, A.A. Pletnev, J. Am. Chem. Soc. 121 (1999) 3238-3239. DOI:10.1021/ja984086w |
| [6] |
E.D. Bergmann, J. Org. Chem. 21 (1956) 461-464. DOI:10.1021/jo01110a023 |
| [7] |
C. Manning, M.R. McClory, J.J. McCullough, J. Org. Chem. 46 (1981) 919-930. DOI:10.1021/jo00318a018 |
| [8] |
S.V. Gagnier, R.C. Larock, J. Am. Chem. Soc. 125 (2003) 4804-4807. DOI:10.1021/ja0212009 |
| [9] |
T. Miura, M. Murakami, Org. Lett. 7 (2005) 3339-3341. DOI:10.1021/ol051249u |
| [10] |
H. Tsukamoto, Y. Kondo, Org. Lett. 9 (2007) 4227-4230. DOI:10.1021/ol701776m |
| [11] |
C.C. Liu, R.P. Korivi, C.H. Cheng, Chem.-Eur. J. 14 (2008) 9503-9506. DOI:10.1002/chem.200801457 |
| [12] |
J. Feng, G. Lu, M. Lv, C. Cai, J. Org. Chem. 79 (2014) 10561-10567. DOI:10.1021/jo501444g |
| [13] |
B.J. Li, H.Y. Wang, Q.L. Zhu, Z.J. Shi, Angew. Chem. Int. Ed. 51 (2012) 3948-3952. |
| [14] |
P. Zhao, F. Wang, K. Han, X. Li, Org. Lett. 14 (2012) 5506-5509. DOI:10.1021/ol302594w |
| [15] |
S. Chen, J. Yu, Y. Jiang, F. Chen, J. Cheng, Org. Lett. 15 (2013) 4754-4757. DOI:10.1021/ol4021145 |
| [16] |
Z. Qi, M. Wang, X. Li, Org. Lett. 15 (2013) 5440-5443. DOI:10.1021/ol4025309 |
| [17] |
X. Yan, S. Zou, P. Zhao, C. Xi, Chem. Commun. 50 (2014) 2775-2777. DOI:10.1039/C4CC00088A |
| [18] |
P. Zhao, Y. Liu, C. Xi, Org. Lett. 17 (2015) 4388-4391. DOI:10.1021/acs.orglett.5b02201 |
| [19] |
N. Lv, Z. Chen, Y. Liu, Z. Liu, Y. Zhang, Org. Lett. 19 (2017) 2588-2591. DOI:10.1021/acs.orglett.7b00906 |
| [20] |
L.H. Lu, S.J. Zhou, W.B. He, et al., Org. Biomol. Chem. 16 (2018) 9064-9068. DOI:10.1039/C8OB02368A |
| [21] |
Y. Zhao, W. Xia, Chem. Soc. Rev. 47 (2018) 2591-2608. DOI:10.1039/C7CS00572E |
| [22] |
M. Yan, J.C. Lo, J.T. Edwards, P.S. Baran, J. Am. Chem. Soc. 138 (2016) 12692-12714. DOI:10.1021/jacs.6b08856 |
| [23] |
J. Wang, K. Sun, X. Chen, et al., Org. Lett. 21 (2019) 1863-1867. DOI:10.1021/acs.orglett.9b00465 |
| [24] |
H. Hu, X. Chen, K. Sun, et al., Org. Chem. Front. 5 (2018) 2925-2929. DOI:10.1039/C8QO00882E |
| [25] |
H. Hu, X. Chen, K. Sun, et al., Org. Lett. 20 (2018) 6157-6160. DOI:10.1021/acs.orglett.8b02627 |
| [26] |
L. Kong, Y. Zhou, F. Luo, G. Zhu, Chin. J. Org. Chem. 38 (2018) 2858-2865. DOI:10.6023/cjoc201805061 |
| [27] |
J.R. Chen, X.Q. Hu, L.Q. Lu, W.J. Xiao, Chem. Soc. Rev. 45 (2016) 2044-2056. DOI:10.1039/C5CS00655D |
| [28] |
K. Jia, Y. Chen, Chem. Commun. 54 (2018) 6105-6112. DOI:10.1039/C8CC02642D |
| [29] |
K. Sun, S.J. Li, X. Chen, et al., Chem. Commun. 55 (2019) 2861-2864. DOI:10.1039/C8CC10243K |
| [30] |
L.Y. Xie, S. Peng, F. Liu, et al., Adv. Synth. Catal. 360 (2018) 4259-4264. |
| [31] |
W. Mai, J. Wang, L. Yang, et al., Chin. J. Org. Chem 34 (2014) 1958-1965. DOI:10.6023/cjoc201405006 |
| [32] |
J. Zhu, W.C. Yang, X.D. Wang, L. Wu, Adv. Synth. Catal. 360 (2018) 386-400. |
| [33] |
M.H. Huang, W.J. Hao, B. Jiang, Chem.-Asian J. 13 (2018) 2958-2977. |
| [34] |
H. Yi, G. Zhang, H. Wang, et al., Chem. Rev. 117 (2017) 9016-9085. DOI:10.1021/acs.chemrev.6b00620 |
| [35] |
M.H. Huang, W.J. Hao, G. Li, S.J. Tu, B. Jiang, Chem. Commun. 54 (2018) 10791-10811. DOI:10.1039/C8CC04618B |
| [36] |
G.P. Yang, X. Wu, B. Yu, C.W. Hu, ACS Sustainable Chem. Eng. 7 (2019) 3727-3732. DOI:10.1021/acssuschemeng.8b06445 |
| [37] |
C. Wu, H.J. Xiao, S.W. Wang, et al., ACS Sustainable Chem. Eng. 7 (2019) 2169-2175. DOI:10.1021/acssuschemeng.8b04877 |
| [38] |
B. Yu, B. Zou, C.W. Hu, J. CO2 Util 26 (2018) 314-322. DOI:10.1016/j.jcou.2018.05.021 |
| [39] |
G.P. Yang, X. He, B. Yu, C.W. Hu, Appl. Organomet. Chem. 32 (2018) e4532. |
| [40] |
G.P. Yang, D. Dilixiati, T. Yang, et al., Appl. Organomet. Chem. 32 (2018) e4450. |
| [41] |
L.Y. Xie, S. Peng, L.L. Jiang, et al., Org. Chem. Front. 6 (2019) 167-171. DOI:10.1039/C8QO01128A |
| [42] |
X. Feng, T. Yang, X. He, B. Yu, C.W. Hu, Appl. Organomet. Chem. 32 (2018) e4314. DOI:10.1002/aoc.4314 |
| [43] |
K.J. Liu, S. Jiang, L.H. Lu, et al., Green Chem. 20 (2018) 3038-3043. DOI:10.1039/C8GC00223A |
| [44] |
K. Wang, L. Yang, W. Zhao, et al., Green Chem. 19 (2017) 1949-1957. DOI:10.1039/C7GC00219J |
| [45] |
K. Okamoto, T. Shimbayashi, E. Tamura, K. Ohe, Org. Lett. 17 (2015) 5843-5845. DOI:10.1021/acs.orglett.5b03065 |
| [46] |
W. Sun, Y. Wang, X. Wu, X. Yao, Green Chem. 15 (2013) 2356-2360. DOI:10.1039/c3gc40980e |
| [47] |
K. Luo, W.C. Yang, L. Wu, Asian J. Org. Chem. 6 (2017) 350-367. |
| [48] |
M.H. Muhammad, X.L. Chen, B. Yu, L.B. Qu, Y.F. Zhao, Pure Appl. Chem. 91 (2019) 33-41. DOI:10.1515/pac-2018-0906 |
| [49] |
R. Li, X. Chen, S. Wei, et al., Adv. Synth. Catal. 360 (2018) 4807-4813. DOI:10.1002/adsc.201801122 |
| [50] |
X.L. Chen, X. Li, L.B. Qu, et al., J. Org. Chem. 79 (2014) 8407-8416. DOI:10.1021/jo501791n |
| [51] |
X. Wo, P. Xie, W. Fu, et al., Chem. Commun. 54 (2018) 11132-11135. DOI:10.1039/C8CC06530F |
| [52] |
P. Xie, J. Fan, Y. Liu, et al., Org. Lett. 20 (2018) 3341-3344. DOI:10.1021/acs.orglett.8b01173 |
| [53] |
Y. Liu, X.L. Chen, F.L. Zeng, et al., J. Org. Chem. 83 (2018) 11727-11735. DOI:10.1021/acs.joc.8b01657 |
| [54] |
X. Chen, X. Li, X.L. Chen, et al., Chem. Commun. 51 (2015) 3846-3849. DOI:10.1039/C4CC10312B |
| [55] |
P. Xie, J. Wang, J. Fan, et al., Green Chem. 19 (2017) 2135-2139. DOI:10.1039/C7GC00882A |
| [56] |
Z.H. Yang, H.R. Tan, J.N. Zhu, J. Zheng, S.Y. Zhao, Adv. Synth. Catal. 360 (2018) 1523-1528. |
| [57] |
Y. Yang, C. Qu, X. Chen, et al., Org. Lett. 19 (2017) 5864-5867. DOI:10.1021/acs.orglett.7b02852 |
| [58] |
Y.M. Li, S.D. Yang, Synlett 24 (2013) 1739-1744. DOI:10.1055/s-00000083 |
| [59] |
Y. Gao, G. Tang, Y. Zhao, Chin. J. Org. Chem 38 (2018) 62-74. DOI:10.6023/cjoc201708023 |
| [60] |
H.F. Qian, C.K. Li, Z.H. Zhou, et al., Org. Lett. 20 (2018) 5947-5951. DOI:10.1021/acs.orglett.8b02639 |
| [61] |
J. Sun, J.K. Qiu, Y.N. Wu, et al., Org. Lett. 19 (2017) 754-757. DOI:10.1021/acs.orglett.6b03546 |
| [62] |
X.Q. Pan, J.P. Zou, G.L. Zhang, W. Zhang, Chem. Commun. 46 (2010) 1721-1723. DOI:10.1039/b925951a |
| [63] |
J. Zhou, G.L. Zhang, J.P. Zou, W. Zhang, Eur. J. Org. Chem. (2011) 3412-34152011. |
| [64] |
C. Pan, B. Huang, W. Hu, X. Feng, J.T. Yu, J. Org. Chem. 81 (2016) 2087-2093. DOI:10.1021/acs.joc.6b00072 |
| [65] |
Y.K. Song, P.C. Qian, F. Chen, C.L. Deng, X.G. Zhang, Tetrahedron 72 (2016) 7589-7593. DOI:10.1016/j.tet.2016.10.013 |
| [66] |
Z. Yan, J. Xie, C. Zhu, Adv. Synth. Catal. 359 (2017) 4153-4157. |
| [67] |
C. Jing, X. Chen, K. Sun, et al., Org. Lett. 21 (2019) 486-489. DOI:10.1021/acs.orglett.8b03768 |
| [68] |
Y. Zhang, S. Ye, M. Ji, et al., J. Org. Chem. 82 (2017) 6811-6818. DOI:10.1021/acs.joc.7b00964 |
| [69] |
S.B. Nagode, A.K. Chaturvedi, N. Rastogi, Asian J. Org. Chem. 6 (2017) 453-457. DOI:10.1002/ajoc.201600549 |
| [70] |
Y. Li, Y. Lu, R. Mao, Z. Li, J. Wu, Org. Chem. Front. 4 (2017) 1745-1750. DOI:10.1039/C7QO00279C |
| [71] |
F.L. Yang, S.K. Tian, Tetrahedron Lett. 58 (2017) 487-504. DOI:10.1016/j.tetlet.2016.12.058 |
| [72] |
G. Qiu, K. Zhou, J. Wu, Chem. Commun. 54 (2018) 12561-12569. DOI:10.1039/C8CC07434H |
| [73] |
J. Wen, W. Shi, F. Zhang, et al., Org. Lett. 19 (2017) 3131-3134. DOI:10.1021/acs.orglett.7b01256 |
| [74] |
X.T. Zhu, Q. Zhao, F. Liu, et al., Chem. Commun. 53 (2017) 6828-6831. DOI:10.1039/C7CC01666B |
| [75] |
X.T. Zhu, T.S. Zhang, Q. Zhao, et al., Chem.-Asian J. 13 (2018) 1157-1164. |
| [76] |
K. Sun, X.L. Chen, X. Li, et al., Chem. Commun. 51 (2015) 12111-12114. DOI:10.1039/C5CC04484G |
| [77] |
F. Liu, J.Y. Wang, P. Zhou, et al., Angew. Chem. Int. Ed. 56 (2017) 15570-15574. DOI:10.1002/anie.201707615 |
| [78] |
W. Li, G. Yin, L. Huang, et al., Green Chem. 18 (2016) 4879-4883. DOI:10.1039/C6GC01196A |
| [79] |
L.Y. Xie, S. Peng, F. Liu, et al., Org. Chem. Front. 5 (2018) 2604-2609. DOI:10.1039/C8QO00661J |
| [80] |
L.Y. Xie, Y.J. Li, J. Qu, et al., Green Chem. 19 (2017) 5642-5646. DOI:10.1039/C7GC02304A |
| [81] |
X.T. Zhu, Q.L. Lu, X. Wang, et al., J. Org. Chem. 83 (2018) 9890-9901. DOI:10.1021/acs.joc.8b01343 |
| [82] |
B. Zhou, W. Chen, Y. Yang, et al., Org. Biomol. Chem. 16 (2018) 7959-7963. DOI:10.1039/C8OB02288G |
| [83] |
K. Sun, X.L. Chen, S.J. Li, et al., J. Org. Chem. 83 (2018) 14419-14430. DOI:10.1021/acs.joc.8b02175 |
| [84] |
X.S. Zhang, J.Y. Jiao, X.H. Zhang, B.L. Hu, X.G. Zhang, J. Org. Chem. 81 (2016) 5710-5716. DOI:10.1021/acs.joc.6b00762 |
| [85] |
Z.J. Shen, Y.N. Wu, C.L. He, et al., Chem. Commun. 54 (2018) 445-448. DOI:10.1039/C7CC08516H |
| [86] |
Z.J. Shen, H.N. Shi, W.J. Hao, S.J. Tu, B. Jiang, Chem. Commun. 54 (2018) 11542-11545. DOI:10.1039/C8CC06086J |
| [87] |
S.K. Pagire, P. Kreitmeier, O. Reiser, Angew. Chem. Int. Ed. 56 (2017) 10928-10932. DOI:10.1002/anie.201702953 |