 2019, Vol. 30
2019, Vol. 30 


Gold, as a heterogeneous catalytic material, has attracted extremely intense interests since Haruta and Hutchings and their co-workers discovered that nano catalysts made from such inert metal can exhibit unique catalytic performance under mild conditions in wide range of applications [1-3]. A lot of experimental and theoretical studies have been conducted to illuminate the origins of the high activity and selectivity of Au based catalysts, in particular the Au nanoparticles or nanorods supported at reducible metal oxides for low-temperature catalytic oxidation of CO and hydrocarbons [4-10]. Various issues including the size effects of nanoparticles, the nature of the metal oxide support and surface adsorption properties have been extensively studied [3]. For example, several studies suggested that, at the low temperature of 90 K, surface adsorbed molecular oxygen is the active species for catalytic CO oxidation at Au nanoparticles supported on the reducible metal oxides [11, 12]; while it was recently found that the oxygen atom can react with CO in the temperature range from 65 K to 250 K [13, 14]. Nevertheless, the nature of adsorbed oxygen species on the Au surface is still unclear, and it is therefore very difficult to thoroughly understand the unique activities of Au based catalysts.
In fact, despite considerable efforts, even some relatively "simple" gold systems remain not well understood in theoretical and experimental studies. For example, Fajin et al. have studied the adsorption of atomic oxygen on the stepped Au(321) surface in detail, and they found that oxygen atoms prefer to interact with surface cavities at steps [15, 16]. On the other hand, Liu et al. proposed that the most stable site for atomic oxygen on stepped Au surfaces is the bridge site of step edges [17].
In addition, the surface structures of some Au facets also remain elusive. For example, the original Au(100) surface should give a square lattice, but experimental measurements often illustrated a close-packed hexagonal layer on top of the square substrate. Some early low-energy electron-diffraction (LEED) and He-scattering studies of this surface indicated a (1 × 5) reconstruction [18, 19]. Several LEED measurements also resolved the splitting in the LEED spots, and suggested a (20 × 5) rather than a (1 × 5) superstructure [20]. Other LEED studies further suggested a larger c (26 × 68) unit cell [21, 22], while even more complex reconstructed structures were found by using scanning tunneling microscopy (STM) [23]. Therefore, it may still need to clarify the general rules that guide the reconstruction of such Au surfaces.
In this work, in order to more thoroughly understand the structural properties of the Au(100) surface and its interaction with reactive species, we performed systematic simulations by using density functional theory (DFT) calculations which were also aided by the genetic algorithm (GA). It needs to be mentioned that early experiments found that adsorbed atomic oxygen could extract Au atoms from the Au(111) substrate [24]. Shi et al. have also predicted that the energetically most favorable configuration of oxygen on Au(111) is a "surface oxide-like" structure that has oxygen being incorporated in its structure [25]. So it is also interesting to learn if oxygen adsorption caused new phases can also occur on the Au (100).
Ingeneral, the resultsof the currentwork show that atomicoxygen adsorption can indeed induce a contracted hexagonal type reconstruction at Au(100). At the same time, the large set of surface structures located through GA aided DFT simulations helped build a formation pathway of the reconstructed surface. Furthermore, CO oxidation at such reconstructedAu(100) phase containingoxygenwas also calculated and it was found to be able to occur rather favorably.
In order to thoroughly exam the potential energy surface (PES) of surface structures, we conducted GA aided DFT calculations by using the USPEX code [26, 27] and the Vienna ab initio simulation package (VASP) [28, 29].
Specifically, USPEX is regarded as a global optimization method, which can produce unexpected structures. The first generation of structures were produced randomly, and the subsequent generations were obtained by using heredity (50%), atom permutation (20%), and softmutation (10%), while others were generated randomly (20%). They are also the typical parameters for USPEX calculations, which are known to have rather high efficiencies.
At the same time, the VASP code was used for the first-principles calculations. The exchange-correlation energy was described by generalized gradient approximation (GGA) in the form of Perdew-Burke-Ernzerhof (PBE) [30]. Previous studies have shown that the GGA-PBE functional can provide a reliable description of the structures and energetics of Au surfaces [31-33]. The interactions between the ions and the electrons were described by the projector augmented wave (PAW) method [34] with a cutoff energy of 400 eV.
A periodic slab model containing four atomic layers was used to simulate the Au(100) surface, which was extended in a (3 × 3) supercell, and a Monkhorst-Pack grid of (3 × 3 × 1) k-point mesh was used. Each slab was separated by a vacuum region of approximately 15 Å. In the structural optimization, we relaxed the atoms in the surface (the top one layer) and buffer regions (the middle two layers), while the other atoms (the bottom one layer) were fixed, until the residual forces were below 0.05 eV/Å.
The adsorption energies for small molecules were calculated with the following equation:

|
where EA/Sub is the total energy of the interacting system containing adsorbate A at the gold substrate (A = CO, O2, etc.), ESub is the energy of the gold substrate, and EA is the energy of the gas-phase molecule, respectively.
Gold is rather inert toward oxygen. Accordingly, once being formed at a gold surface, oxygen atoms would be very unstable and reactive. However, formation of atomic oxygen via direct O2 dissociation is difficult at Au(100) because of a high activation barrier [35-37]. In addition, active oxygen species other than atomic oxygen may also induce oxidation reactions at Au. For example, the reaction between adsorbed O2 and CO through intermediate OC–OO complex (OC + O2 → OC-OO → CO2 + O) has been proposed at Au based catalysts [38, 39], and both the product (CO2) and very active O atom that would further oxidize another CO can occur.
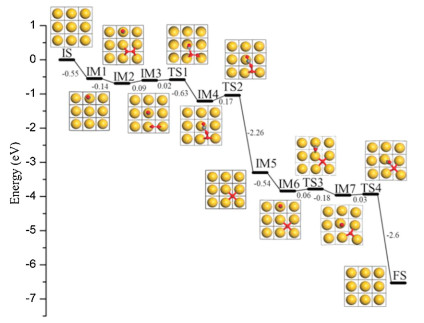
In this work, we mainly studied CO oxidation on the Au(100) surface. Firstly, we calculated the energy profile of CO catalytic oxidation on the Au(100) surface as a hard template (Fig. 1). The clean surface was considered in the initial state (IS), and the CO and O2 were adsorbed successively with the adsorption energies of 0.55 eV (IM1) and 0.14 eV (IM2), respectively, which are well consistent with previous calculation results [40]. Then, the coadsorbed CO and O2 can move toward each other (IM3) and react through the transition state (TS1) to form the OC-OO species (IM4) with the calculated energy barrier of 0.02 eV only. The O——O bond in this OC-OO species can further break with the energy barrier of 0.17 eV (TS2), and afterwards, the CO2 can form and desorb with a negligible desorption energy, leaving the surface with an adsorbed atomic oxygen on the four-fold hollow site (IM5). In the end, the surface atomic oxygen can continue to react with another adsorbed CO (IM6) when they move toward each other (TS3 and IM7), and the energy barrier calculated for the last step of the reaction is 0.03 eV only (TS4). In general, one can see that the calculated activation energies for the total reactions between CO and O2 are quite low on the Au(100) surface, though the O2 adsorption is also very weak on the Au(100) surface, which may affect the overall reactivities.

|
Download:
|
| Fig. 1. Calculated energy profile of catalytic CO oxidation on the Au(100) surface through reactions with adsorbed molecular O2 and atomic O. The Au atoms are in gold, O in red and C in grey, and these notations are used throughout the whole manuscript. | |
{kind=link}
It needs to be mentioned that the above reaction processes between these simple adsorbents completely ignored whether the Au catalyst could be affected by the reaction [41, 42]. Due to its unique structural flexibility [43] and the influence of adsorbed species, the Au catalyst is actually very likely to undergo surface reconstruction. Accordingly, we used the global GA method to study the effect of the adsorption on the gold surface, which has been successfully applied to predict the structures of crystals, clusters, reconstructed surfaces and polymers [44].
In this work, the possible structures of the Au(100) surface containing different adsorbates such as two atomic oxygen or CO in one surface cell have been systematically located by the GA aided total energy DFT calculations. In general, the results showed that CO cannot induce Au surface reconstruction, but adsorbed oxygen species can drastically change the surface morphologies. A large number of stable structures of the Au(100) surface containing two adsorbed oxygen atoms (as well as the clean one and those with one singe O) were determined in this work (Fig. 2). The one with the lowest total energy shows that the two oxygen atoms are adsorbed separately (Fig. 2f), indicating that the dissociative adsorption of one O2 molecule is preferred. More importantly, one can also see from the figure that the substrate Au(100) surface undergoes complete rearrangement for the top layer atoms to achieve the hexagonal pattern, yet the other layers still remain the original pattern. Compared with the bulk truncated Au(100), the reconstructed Au(100) substrate appears to occur through the way that one gold chain among every three slides by half lattice unit, and at the same time, the other two nearby shrink toward the sliding chain (Figs. 2a and d). In addition, we can further find that at such hexagonal type reconstructed surface, the surface Au atom that directly bonds with the two O is lifted out of the surface plane, and as the result, a protruding linear O-Au-O species occurs on the surface (Fig. 2f).

|
Download:
|
| Fig. 2. Calculated most stable structures of unreconstructed and reconstructed (a, d) Au(100) and the surfaces with (b, e) one and (c, f) two O in one surface cell. | |
{kind=link}
According to the above results, two co-adsorbed oxygen atoms at Au(100) can induce the hexagonal type surface reconstruction. In fact, our calculations may also determine the coverage effect of oxygen on the occurrence of this reconstruction. The most stable structures of the unreconstructed and reconstructed Au(100) surface containing adsorbed oxygen under different coverages of 0, 1/9 and 2/9 ML (the number of adsorbed O atoms with respect to surface Au in one surface cell) obtained from GA aided DFT calculations are illustrated in Fig. 2, and the calculated energy differences are 0.07 eV, 0.03 eV and -0.07 eV, respectively. It can be clearly seen that when oxygen coverage gradually increases, the energy difference gradually decreases, and the Au(100) surface thermodynamically prefers to reconstruct in the hexagonal way when the O coverage reaches 2/9 ML. These results indicate that adsorptions of oxygen only with the coverages high enough can make the Au(100) surface undergo reconstruction.
For the Au(100) surface with two O in a 3 × 3 surface cell (2/9 ML), the GA aided DFT calculations can not only help determine the most stable adsorption structures, the obtained meta-stable structures can even help build the whole reconstruction process induced by the adsorption. First, we took the clean and unreconstructed Au(100) as the starting point. Then, we considered the obtained structures corresponding to the adsorption of O2 and its subsequent dissociation. From the rational arrangement on the basis of calculated energetics and structures for the intermediates [45], we can construct the PES of the evolution pathway for the reconstruction. In addition, we could also locate the transition states between these intermediate states to make the PES more complete.
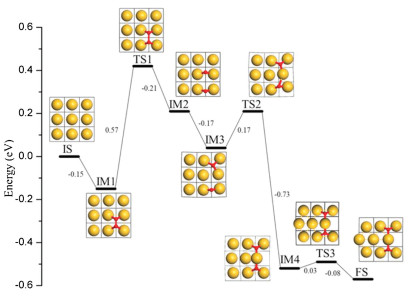
The PES determined in the above way is illustrated in Fig. 3. From the calculated evolution pathway, one can obviously see that the activation energy for O2 dissociation (0.57 eV, TS1) is quite small, though it is still higher than the calculated O2 adsorption energy (0.15 eV, IM1), which suggests that formation of atomic oxygen via direct O2 dissociation is indeed difficult at Au(100) and gold is rather inert toward oxygen [32, 46]. This is different from some other transition metals, such as Pt, on which direct O2 dissociation can readily occur and generate adsorbed O atoms to oxidize CO [9, 47]. However, it is quite surprising to see that once the two atomic O from O2 dissociation co-exist at Au(100) (IM2 and IM3), they can induce the hexagonal type reconstruction of the Au (100) substrate through a two-step process with very small energy barriers (0.17 eV and 0.03 eV, respectively). Specifically, for the first step, the two co-adsorbed O move from their own adsorption sites (IM3) to bind with the same Au atom (TS2) and cause it to shift by nearly half unit (IM4). Then, the nearby surface Au atoms shift further (TS3) to form the final reconstructed surface (FS). From the calculated energetics (Fig. 3), one can also clearly see that such hexagonal type reconstruction as well as the formation of the linear O-Au-O structure on Au(100) surface can greatly stabilize the whole surface as the reconstructed surface (FS) is ~0.4 eV more stable than that with the adsorbed molecular O2 (IM1).

|
Download:
|
| Fig. 3. Calculated energy profile of Au(100) reconstruction induced by O2 adsorption and dissociation. | |
{kind=link}
The results of GA aided DFT calculations in this work show that oxygen species can induce Au surface reconstruction. However, it should be noted that our calculated results also indicate that O2 dissociation is difficult to occur. Therefore, it is very likely that O2 may still involve in other surface processes rather than dissociation. We expected that the reaction between CO and O2 occur firstly on the surface of unreconstructed Au(100) and combined the PES in Fig. 1 and Fig. 3 to plot a new one in Fig. 4a, from which one can see that the CO and O2 can adsorb in the same surface cell (IM1 and IM2) and migrate toward each other (IM3) to achieve CO2 formation (IM5) through the occurrence of the OC-OO intermediate (IM4) with relatively low reaction barriers (TS1 and TS2). The remaining O after the direct OCOO dissociation and CO2 formation may react with another CO as show in Fig. 1. However, under the conditions of low CO concentration, such adsorbed atomic O may also accumulate to induce the surface reconstruction. So, we considered the accumulation of the O and calculated the coadsorbed two O in the same surface cell. This process corresponds to the evolution of IM5 to IM6 and their energy difference is due to the competitive adsorption of the two O. Afterward, the surface may follow the same reconstruction process as shown in Fig. 3 to form the hexagonal type reconstructed Au(100) (IM6 to FS).

|
Download:
|
| Fig. 4. Calculated energy profile of CO oxidation on the unreconstructed Au(100) surface and the induced surface reconstruction (a), and that of CO oxidation on the linear O-Au-O structure of the reconstructed Au(100) (b). | |
{kind=link}
In order to understand the reactivity of the reconstructed Au (100), especially that of the unique O-Au-O species at the reconstructed surface, we then calculated the processes including CO adsorption and oxidation. Fig. 4b shows the calculated energy profile of these processes. The calculated adsorption energy of CO on the linear O-Au-O structure is 0.53 eV (IM1), and the energy barrier of CO reaction with the O of O-Au-O is 0.56 eV only (TS1), which is nearly identical to its adsorption energy. It needs to be mentioned that after CO2 formation, the surface may end up with the single O being adsorbed at either the hexagonal type reconstructed surface or that recovering the original Au(100) configuration, both of which have almost the same stability (IM2). Then, an extra CO may continue to adsorb (IM3) and react with the remaining O with very small barriers (TS2). Finally, the clean surfaces can occur and the unreconstructed Au(100) is relatively more stable than the reconstructed one as we have explained in the above.
In this work, we have studied the adsorptions of oxygen species on the Au(100) surface and their influence on the structures of the Au(100) substrate as well as the surface activities in catalytic oxidation of CO by using the GA aided total energy DFT calculations. The results show that the interactions of oxygen atoms and Au(100) can affect the surface morphology by inducing the hexagonal type reconstruction to the surface layer and forming a lifted O-Au-O species. Such reconstructed partially oxidized Au surface is significantly more stable than that involving the molecularly or dissociatively adsorbed O2 at the pristine Au(100). Moreover, this reconstructed Au(100) surface is also highly reactive for CO oxidation as the oxygen in the O-Au-O species can directly react with adsorbed CO through a Mars-van Krevlen type mechanism, which is completely different from those for CO oxidation through reaction with adsorbed O2 or O at the unreconstructed Au(100) as a hard template. This work reveals the kinetic nature of the Au surface that it can undergo drastic reconstruction under reaction conditions and maintain high catalytic activities. This work also demonstrates that the genetic algorithm and other intelligent algorithm methods can largely improve the capacity of first-principles calculations to more thoroughly sample the PES of structures and chemical processes in heterogeneous catalysis [48-51].
AcknowledgmentsThis work was supported by National Key R&D Program of China (No. 2018YFA0208602), National Natural Science Foundation of China (Nos. 21421004, 21825301, 21573067, 91545103) and Program of Shanghai Academic Research Leader (No. 17XD1401400). The authors also thank the National Super Computing Center in Jinan for computing time.
| [1] |
M. Haruta, Chem. Rec. 3 (2003) 75-87. DOI:10.1002/(ISSN)1528-0691 |
| [2] |
M. Haruta, M. Daté, Appl. Catal. A 222 (2001) 427-437. DOI:10.1016/S0926-860X(01)00847-X |
| [3] |
R. Meyer, C. Lemire, S.K. Shaikhutdinov, H.J. Freund, Gold Bull. 37 (2004) 72-124. DOI:10.1007/BF03215519 |
| [4] |
M. Haruta, N. Yamada, T. Kobayashi, S. Iijima, J. Catal. 115 (1989) 301-309. DOI:10.1016/0021-9517(89)90034-1 |
| [5] |
T. Hayashi, K. Tanaka, M. Haruta, J. Catal. 178 (1998) 566-575. DOI:10.1006/jcat.1998.2157 |
| [6] |
M. Haruta, Catal. Today 36 (1997) 153-166. DOI:10.1016/S0920-5861(96)00208-8 |
| [7] |
M.M. Schubert, S. Hackenberg, van Veen A.C., et al., J. Catal. 197 (2001) 113-122. DOI:10.1006/jcat.2000.3069 |
| [8] |
C.K. Costello, M.C. Kung, H.S. Oh, Y. Wang, H.H. Kung, Appl. Catal. A 232 (2002) 159-168. DOI:10.1016/S0926-860X(02)00092-3 |
| [9] |
Z.P. Liu, X.Q. Gong, J. Kohanoff, C. Sanchez, P. Hu, Phys. Rev. Lett. 91 (2003) 266102. DOI:10.1103/PhysRevLett.91.266102 |
| [10] |
I.N. Remediakis, N. Lopez, J.K. Nørskov, Angew. Chem. Int. Ed. 44 (2005) 1824-1826. DOI:10.1002/anie.200461699 |
| [11] |
F. Boccuzzi, A. Chiorino, S. Tsubota, M. Haruta, J. Phys. Chem. 100 (1996) 3625-3631. DOI:10.1021/jp952259n |
| [12] |
F. Boccuzzi, A. Chiorino, M. Manzoli, Mater. Sci. Eng. C 15 (2001) 215-217. DOI:10.1016/S0928-4931(01)00222-3 |
| [13] |
T.S. Kim, J.D. Stiehl, C.T. Reeves, R.J. Meyer, C.B. Mullins, J. Am. Chem. Soc. 125 (2003) 2018-2019. DOI:10.1021/ja028719p |
| [14] |
J.D. Stiehl, T.S. Kim, C.T. Reeves, R.J.M. And, C.B. Mullins, J. Phys. Chem. B 108 (2004) 7917-7926. DOI:10.1021/jp0496102 |
| [15] |
J. L.C. Fajín, J.R.B. Gomes, J. Phys. Chem. C 111 (2007) 17311-17321. DOI:10.1021/jp073796y |
| [16] |
J.L.C. Fajín, M.N.D.S. Cordeiro, J.R.B. Gomes, J. Phys. Chem. C 112 (2008) 17291-17302. DOI:10.1021/jp8031435 |
| [17] |
Z.P. Liu, P. Hu, A. Alavi, J. Am. Chem. Soc. 124 (2002) 14770. DOI:10.1021/ja0205885 |
| [18] |
D.G. Fedak, N.A. Gjostein, Phys. Rev. Lett 16 (1966) 171-172. DOI:10.1103/PhysRevLett.16.171 |
| [19] |
K.H. Rieder, T. Engel, R.H. Swendsen, M. Manninen, Surf. Sci. 127 (1983) 223-242. DOI:10.1016/0039-6028(83)90415-6 |
| [20] |
D.G. Fedak, N.A. Gjostein, Surf. Sci. 8 (1967) 77-97. DOI:10.1016/0039-6028(67)90074-X |
| [21] |
J.F. Wendelken, D.M. Zehner, Surf. Sci. 71 (1978) 178-184. DOI:10.1016/0039-6028(78)90325-4 |
| [22] |
M.A.V. Hove, R.J. Koestner, P.C. Stair, et al., Surf. Sci. 103 (1981) 189-217. DOI:10.1016/0039-6028(81)90107-2 |
| [23] |
O.K. Binnig, H. Rohrer, C. Gerber, E. Stoll, Surf. Sci. 144 (1984) 321-335. DOI:10.1016/0039-6028(84)90104-3 |
| [24] |
B.K. Min, X. Deng, D. Pinnaduwage, R. Schalek, C.M. Friend, Phys. Rev. B:Condens. Matter Mater. Phys. 72 (2005) 121410. DOI:10.1103/PhysRevB.72.121410 |
| [25] |
H. Shi, C. Stampfl, Phys. Rev. B:Condens. Matter Mater. Phys. 76 (2007) 075327. DOI:10.1103/PhysRevB.76.075327 |
| [26] |
A.R. Oganov, C.W. Glass, J. Chem. Phys. 124 (2006) 201-419. |
| [27] |
A.R. Oganov, A.O. Lyakhov, M. Valle, Acc. Chem. Res. 44 (2011) 227-237. DOI:10.1021/ar1001318 |
| [28] |
G. Kresse, J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys. 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [29] |
G. Kresse, J. Furthmüller, Comp. Mater. Sci. 6 (1996) 15-50. DOI:10.1016/0927-0256(96)00008-0 |
| [30] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [31] |
T.A. Baker, C.M. Friend, E. Kaxiras, J. Phys. Chem. C 113 (2009) 3232-3238. DOI:10.1021/jp806952z |
| [32] |
H. F. Wang, X.Q. Gong, Y.L. Guo, et al., J. Phys. Chem. C 113 (2009) 6124-6131. DOI:10.1021/jp810608c |
| [33] |
L.M.C. Pinto, G. Maia, J. Phys. Chem. C 119 (2015) 8213-8216. DOI:10.1021/acs.jpcc.5b01358 |
| [34] |
P.E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys. 50 (1994) 17953-17979. DOI:10.1103/PhysRevB.50.17953 |
| [35] |
D.H. Parker, B.E. Koel, J. Vac. Sci. Technol. A 8 (1990) 2585-2590. DOI:10.1116/1.576675 |
| [36] |
J.D. Stiehl, T.S. Kim, S.M. McClure, C.B. Mullins, J. Am. Chem. Soc. 126 (2004) 13574-13575. DOI:10.1021/ja046390x |
| [37] |
Y. Xu, M. Mavrikakis, J. Phys. Chem. B 107 (2003) 9298-9307. DOI:10.1021/jp034380x |
| [38] |
M. Okumura, J.M. Coronado, J. Soria, M. Haruta, J.C. Conesa, J. Catal. 203 (2001) 168-174. DOI:10.1006/jcat.2001.3307 |
| [39] |
H. Liu, A.I. Kozlov, A.P. Kozlova, et al., J. Catal. 185 (1999) 252-264. DOI:10.1006/jcat.1999.2517 |
| [40] |
A. Hussain, J. Gracia, J.W. Niemantsverdriet, B.E. Nieuwenhuys, Molecules 16 (2011) 9582-9599. DOI:10.3390/molecules16119582 |
| [41] |
B.B. Blizanac, C.A. Lucas, M.E. Gallagher, et al., J. Phys. Chem. B 108 (2004) 625-634. DOI:10.1021/jp036483l |
| [42] |
J.L.C. Fajín, M.N.D.S. Cordeiro, J.R.B. Gomes, Surf. Sci. 602 (2008) 424-435. DOI:10.1016/j.susc.2007.10.037 |
| [43] |
S.C. Street, A. Rar, J.N. Zhou, W.J. Liu, J.A. Barnard, Chem. Mater. 13 (2001) 3669-3677. DOI:10.1021/cm000981e |
| [44] |
S. Franzen, Chem. Phys. Lett. 381 (2003) 315-321. DOI:10.1016/j.cplett.2003.08.126 |
| [45] |
P. Ding, X. Q. Gong, J. Mol. Model. 22 (2016) 114. DOI:10.1007/s00894-016-2977-1 |
| [46] |
X. Deng, B.K. Min, A. Guloy, C.M. Friend, J. Am. Chem. Soc. 127 (2005) 9267-9270. DOI:10.1021/ja050144j |
| [47] |
X.Q. Gong, Z.P. Liu, R. Raval, P. Hu, J. Am. Chem. Soc. 126 (2004) 8-9. DOI:10.1021/ja030392k |
| [48] |
A.R. Oganov, S. Ono, Nature 430 (2004) 445-448. DOI:10.1038/nature02701 |
| [49] |
Q. Wang, A.R. Oganov, O.D. Feya, Q. Zhu, D. Ma, Phys. Chem. Chem. Phys. 18 (2016) 19549-19556. DOI:10.1039/C6CP01203E |
| [50] |
H.A. Zakaryan, A.G. Kvashnin, A.R. Oganov, Sci. Rep. 7 (2017) 10357. DOI:10.1038/s41598-017-10331-z |
| [51] |
X.H. Yu, A.R. Oganov, I.A. Popov, G.R. Qian, I.A. Boldyrev, Angew. Chem. Int. Ed. 128 (2016) 1731-1735. DOI:10.1002/ange.201508439 |