 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Chemical Oncogenomics, Key Laboratory of Chemical Biology, the Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China;
c National & Local United Engineering Lab for Personalized Anti-tumor Drugs, Shenzhen Kivita Innovative Drug Discovery Institute, the Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China;
d School of Life Science, Tsinghua University, Beijing 100084, China;
e School of Pharmaceutical Sciences, Tsinghua University, Beijing 100084, China
Histone deacetylases (HDACs) are important epigenetic enzymes involved in chromatin remodeling, thereby directly regulating the transcription of various genes by removing the acetyl group of histone lysines [1, 2]. Recent studies indicate that HDAC inhibitors (HDACi) display remarkable therapeutic efficacy against certain cancers [3-5]. Of the various developed HDAC inhibitors [6-9], five have been approved for the treatment of refractory cutaneous T-cell lymphoma (CTCL) (suberoylanilide hydroxamic acid (SAHA) and romidepsin), or peripheral T-cell lymphoma (PTCL) (romidepsin, belinostat, and chidamide), or for use in patients with recurrent multiple myeloma (panobinostat) [10, 11].
Most HDAC inhibitors, including SAHA, belinostat, and panobinostat, share essential pharmacophoric structural features: a cap group interacting with the surface of the enzyme, a zincbinding group (ZBG), such as a hydroxamic acid chelating group, chelating the zinc atom in the HDAC catalytic site, and a linker with the proper length to connect them [12]. Their flexible structureactivity relationship leads to the easy connection of HDAC inhibitors with a variety of other anti-cancer core scaffolds, promoting the extensive development of dual-target HDACi drugs, which possess enhanced anticancer efficacy [7, 13]. Despite the reports on the effects of suppressing tumor cell proliferation of these developed hybrids, their underlying molecular mechanisms require additional study.
In recent decades, significant advances in omics technology have enabled high-throughput monitoring of multiple molecular and biological processes, making it a powerful platform to explore the action mechanisms of drugs [14, 15]. As major carriers and function performers of cellular activities, proteins carry a wealth of biological information [16], allowing proteomics to provide important insights into the mechanism of drug action through a comprehensive analysis of protein alterations [17-19].
Apart from HDAC, DNA methyltransferase (DNMT) is another important epigenetic target in anticancer drug development. Two DNMT inhibitors, 5-azacytidine and decitabine, were approved by FDA in 2004 and 2006, respectively. Previous studies have shown that the combination of DNMTi and HDACi displayed improved potency in inhibiting the development of multiple types of tumor [20-23]. Focusing on the development of and having successfully synthesized multitargeted antitumor agents [24-27], our group recently synthesized a series of hydroxamic acid derivatives as DNMT and HDAC dual target inhibitors. Among them, compound 208 (Fig. 1A) displayed potent HDAC inhibitory activity against HDAC1 and HDAC6 with IC50 values of 2.40 nmol/L and 4.83 nmol/L, respectively, less than that of the reference compound SAHA (HDAC1: IC50 = 11.11 nmol/L; HDAC6: IC50 = 7.76 nmol/L), and exhibited a 44.48% inhibition rate on DNMT1 at 50 μmol/L in the H-3-labeled radioactive methylation assay [28] (Table S1 in Supporting information). The cytotoxicity assay results (Table S2 in Supporting information) revealed that 208 possesses substantial antiproliferative activity against multiple tumor cells at low concentrations and could effectively suppress the proliferation of U937 cells with an IC50 value of 1.30 μmol/L at 72 h.

|
Download:
|
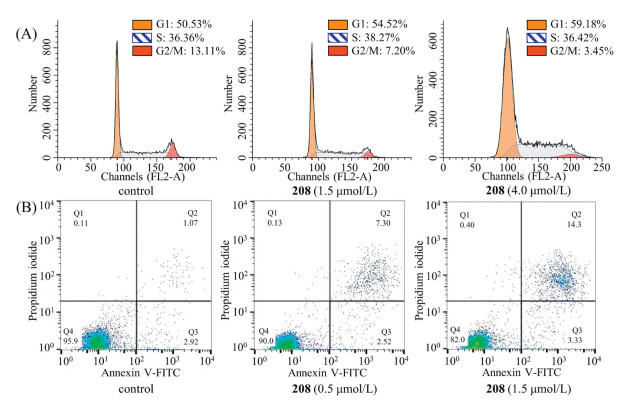
| Fig. 1. (A) Chemical structure of hydroxamic acid derivative 208; (B) 208 induced hyperacetylation of histones H3K9 and H4K8; (C) 208 increased the expression of tumor suppressor protein p16 (U937 cells were treated with indicated concentration of 208 for 12 h). | |
{kind=link}
The inhibition of HDAC increases the level of histone acetylation and consequently turns the chromatin to be loose and active, allowing multiple transcription factors to access to promoters of diverse genes including tumor suppressor genes [27]. As compound 208 possessed potent HDAC inhibitory activity and showed considerable DNMT1 inhibitory potency, we further explored its effect on the acetylation of histones and the expression of relevant proteins in U937 cells via immunoblot analysis. Treated with 208 for 12 h, U937 cells displayed a dosedependent pattern of increased acetylation of histones H3K9 and H4K8 (Fig. 1B), confirming the inhibitory effectiveness of 208 [29]. It has also been reported that some DNMT inhibitors, such as decitabine and SGI-1027, can reactivate the silenced tumor suppressor gene CDKN2A and increase its expression product p16 accordingly [30]. As expected, the results of western blot analysis (Fig. 1C) indicated that the expression of p16 in U937 cells treated with 208 for 12 h was upregulated as the concentration of 208 increased.
P16, also known as multiple tumor suppressor 1, is an important negative feedback regulator of the cell cycle which can inhibit the activity of cyclin-dependent kinase 4 (CDK4) by competing with cyclin to bind CDK4, thereby hindering the cell cycle from G1 to S phase [31]. Therefore, we analyzed cell cycle arrest and apoptosis in U937 cells treated with 208 using flow cytometry. Our cell cycle arrest analyses indicated that U937 cells were markedly arrested in the G1 phase (Fig. 2A) in response to 208 treatment. As the concentration of 208 gradually increased from 0 to 4.0 μmol/L, the percentage of U937 cells at G1 phase increased from 50.53% to 59.18%. Treated with 208 at different concentrations for 48 h, the population of apoptotic U937 cells exhibited a dose-dependent increasing trend (Fig. 2B), suggesting that 208 can trigger apoptosis process to inhibit U937 cell proliferation.

|
Download:
|
| Fig. 2. (A) 208 arrested cell cycle progression at the G1 phase; (B) 208 induced substantial apoptosis in U937 cells. U937 cells were treated for 48 h. | |
{kind=link}
Histone acetylation and DNA methylation affect gene expression by regulating chromatin remodeling, which is involved in all the hallmarks of tumor [21, 32]. To investigate the complex antitumor mechanism of 208 as a dual-target epigenetic inhibitor, including the potential functional mechanisms of 208-induced U937 cell cycle arrest and apoptosis, we conducted label-free quantitative proteomics analysis using Nano LC-MS/MS, profiling the proteome in U937 cells with or without 208 treatment. To explore changes in proteomes at different drug treatment times, we collected cell lysates from 208-treated or control cells after 12 h, 24 h and 48 h as three groups. The concentrations of drug were uniformly set at 3 μmol/L to induce sufficient cellular changes and appropriate levels of cell death based on MTT assay results.
Over 1800 nonredundant quantifiable proteins were identified in U937 cells treated in each group. Only proteins changed over 1.5 folds were selected for further analysis, whose numbers were 343, 307, and 429 after treated for 12 h, 24 h, and 48 h. In comparison, 115 differential proteins of the three groups were overlapping. Additionally, the 12 h treated protein group had 104 differential proteins that were different from those in the other two groups and 63 proteins were unique in the 24 h group, compared with 144 in the 48 h group (Fig. S1 in Supporting information). In short, through effects on epigenetic modifications, 208 differentially modulated the protein expression in treated U937 cells over time.
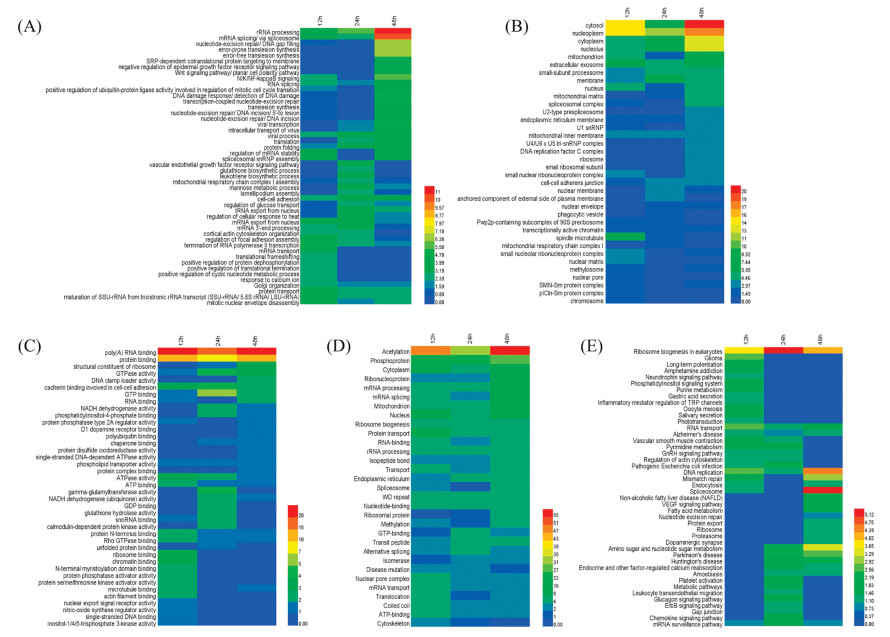
To elucidate the biological functions of these differentially expressed proteins, we analyzed the proteome data sets with the online analysis platform DAVID, classifying them into three enrichment gene ontology (GO) categories accordingly: biological processes, cellular composition, and molecular function. The enrichment analysis results revealed that in the biological processes category (Fig. 3A), significantly enriched proteins were related to termination of RNA polymerase Ⅱ transcription (p = 1.10 × 10-5, 12 h), rRNA processing (p = 7.40 × 10-7, 24 h), mRNA splicing (via spliceosome) (p = 8.40 × 10-11, 48 h), nucleotideexcision repair, DNA gap filling (p = 1.50 × 10-7, 48 h), etc., which are processes relevant to gene transcription, translation, and DNA damage. DNA damage response, detection of DNA damage (p = 4.50 × 10-6, 48 h), was also a highly enriched category, in agreement with previous study that HDAC inhibitors can attenuate the levels of DNA damage response [33]. In the cellular composition analysis (Fig. 3B), mitochondria-associated proteins were significantly enriched by 208 treatment, consistent with the observation that mitochondria are the primary intracellular compartments in response to acetylation-related events [34]. Interestingly, we found that 208 treatment had profound effects on proteins of the extracellular exosome, which are suggested to play a role in cell-to-cell signaling, potentially influencing cancer development [35]. Furthermore, analysis of the molecular function category (Fig. 3C) indicated that these proteins are mostly involved in the binding of poly (A) RNA (p = 1.40 × 10-17, 24 h), protein (p = 1.60 × 10-12, 12 h), and ribosome (p = 1.40 × 10-4, 12 h), and structural constituent of ribosome (p = 1.90 × 10-4, 48 h).

|
Download:
|
| Fig. 3. Enrichment-based clustering analysis for the functional annotation of proteins from U937 cells which were altered by 208 treatment from the DAVID online database: (A) biological processes, (B) cellular composition, (C) molecular function, (D) UP_Keywords, and (E)KEGG pathway.The colors represent -log10p-value of proteinsin each gene ontology category. | |
{kind=link}
We also conducted analyses on the functional category of UP_Keywords and on KEGG pathways for further understanding. The UP_Keywords analysis (Fig. 3D) demonstrated that acetylation and phosphoprotein were highly enriched categories. KEGG pathways categories (Fig. 3E) of ribosome biogenesis in eukaryotes, DNA replication, and mismatch repair, were significantly enriched in response to 208 treatment. The significant enrichment in the ribosome biogenesis pathway appears to be consistent with the proteomics analysis of SAHA [34]. Differential proteins involved in each category are summarized in Table S3 (Supporting information). The protein-protein interaction maps (Fig. S2 in Supporting information) of differential proteins were generated after importing them into the STRING tool and these results are consistent with the biological function enrichment analysis by DAVID enrichment platform. The biological functional analysis helped us to elucidate the effects on biological functions of differential proteins in response to multi-target epigenetic inhibitors.
The G1 phase of the cell cycle is the specific period in which cells make decisions about growth and division. Cyclins and cyclindependent kinases (CDKs), which are modulated by a complex mechanism, are the key controlling factors of cell cycle. We found that proteins within the UP_Keywords category of phosphoprotein, which play an important role in cell cycle control from G1 to S phase, were highly enriched. Among these proteins, we observed that compound 208 substantially reduced the expression of several proteins associated with G1 phase cell cycle arrest and apoptosis, including p85α, MEK, and CDK4. Interestingly, p85α can modulate phosphorylation of MEK through RAC1/PAK pathway [36]. We also noted the effect of 208 on the expression of these proteins by immunoblotting. As shown in Fig. 4A, combined inhibition of HDAC and DNMT by 208 can significantly decrease the expression of CDK4 and its modulators, p85α and MEK, and reduce MEK phosphorylation. Taken together, our data demonstrated that 208 induces cell cycle arrest at G1 phase to inhibit cancer progression through upregulating CDK inhibitor p16 and downregulating cyclin-dependent kinases and their activators (Fig. 4B). These results suggest a new potential strategy of HDAC and DNMT inhibitors in cancer therapy.

|
Download:
|
| Fig. 4. (A) 208 significantly decreased the expression of p85α, MEK, p-MEK, and CDK4 (treated for 12 h). (B) The schematic of the effect of 208 on cell cycle arrest and apoptosis. Green arrows indicate downregulation, the red arrow indicates upregulation, black arrows indicate activation, and blunt ends indicate inhibition. | |
{kind=link}
In this study, we reported the dual target epigenetic inhibitor, 208, which displays potent DNMT1 and HDAC inhibitory activity and exhibits substantial antiproliferative activity against U937 histiocytic lymphoma cells. Our study combined label-free quantitative proteomics with biochemical assays and bioinformatic analysis to investigate the proteome pattern in 208 treated U937 cells. The results indicate that compound 208, which alters epigenetic modification, could subsequently affect the entire proteome profile and may regulate the expression of key proteins in U937 cells, influencing multiple pathways to induce cell cycle arrest and apoptosis. These results advance the understanding of the underlying pharmacological mechanisms of HDAC and DNMT inhibitors.
AcknowledgmentsWe appreciate the financial supports from Shenzhen Development and Reform Committee (No. 20151961), China Postdoctoral Science Foundation (No. 2018M631825) and Department of Science and Technology of Guangdong Province (No. 2017B030314083).
Appendix A. Supplementary dataSupplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.03.029.
| [1] |
M.A. Dawson, T. Kouzarides, Cell 150 (2012) 12-27. DOI:10.1016/j.cell.2012.06.013 |
| [2] |
M.D. Shahbazian, M. Grunstein, Annu. Rev. Biochem. 76 (2007) 75-100. DOI:10.1146/annurev.biochem.76.052705.162114 |
| [3] |
L. Wang, H. Li, Y. Ren, et al., Cell Death Dis. 7 (2016) 2063. DOI:10.1038/cddis.2015.328 |
| [4] |
K. Xue, J.J. Gu, Q. Zhang, et al., J. Cancer Res. Clin. Oncol. 142 (2016) 379-387. DOI:10.1007/s00432-015-2026-y |
| [5] |
B. Kumar, A. Yadav, J.C. Lang, et al., Genes Cancer 6 (2015) 169-181. |
| [6] |
F. Thaler, L. Moretti, R. Amici, et al., Eur. J. Med. Chem. 108 (2016) 53-67. DOI:10.1016/j.ejmech.2015.11.010 |
| [7] |
J. Roche, P. Bertrand, Eur. J. Med. Chem. 121 (2016) 451-483. DOI:10.1016/j.ejmech.2016.05.047 |
| [8] |
C.J. Gong, A.H. Gao, Y.M. Zhang, et al., Eur. J. Med. Chem. 112 (2016) 81-90. DOI:10.1016/j.ejmech.2016.02.003 |
| [9] |
A.C. West, R.W. Johnstone, J. Clin. Invest. 124 (2014) 30-39. DOI:10.1172/JCI69738 |
| [10] |
S. Yoon, G.H. Eom, Chonnam Med. J. 52 (2016) 1-11. DOI:10.4068/cmj.2016.52.1.1 |
| [11] |
Y. Shi, B. Jia, W. Xu, et al., J. Hematol. Oncol. 10 (2017) 69. DOI:10.1186/s13045-017-0439-6 |
| [12] |
K.J. Falkenberg, R.W. Johnstone, Nat. Rev. Drug Discov. 13 (2014) 673-691. DOI:10.1038/nrd4360 |
| [13] |
H.M. Hesham, D.S. Lasheen, K.A.M. Abouzid, Med. Res. Rev. 38 (2018) 2058-2109. DOI:10.1002/med.2018.38.issue-6 |
| [14] |
C. Manzoni, D.A. Kia, J. Vandrovcova, et al., Briefings Bioinf. 19 (2018) 286-302. DOI:10.1093/bib/bbw114 |
| [15] |
V.G. Anania, J.R. Lill, Proteomics:Clin. Appl. 9 (2015) 671-683. DOI:10.1002/prca.v9.7-8 |
| [16] |
R. Aebersold, M. Mann, Nature 537 (2016) 347-355. DOI:10.1038/nature19949 |
| [17] |
Y. Zhao, S. Miriyala, L. Miao, et al., Free Rad. Biol. Med. 72 (2014) 55-65. DOI:10.1016/j.freeradbiomed.2014.03.001 |
| [18] |
J. Lin, X.D. Li, Chin. Chem. Lett. 29 (2018) 1051-1057. DOI:10.1016/j.cclet.2018.05.017 |
| [19] |
M. Breker, M. Schuldiner, Nat. Rev. Mol. Cell Biol. 15 (2014) 453-464. DOI:10.1038/nrm3821 |
| [20] |
R. Pathania, S. Ramachandran, G. Mariappan, et al., Cancer Res. 76 (2016) 3224-3235. DOI:10.1158/0008-5472.CAN-15-2249 |
| [21] |
N. Azad, C.A. Zahnow, C.M. Rudin, S.B. Baylin, Nat. Rev. Clin. Oncol. 10 (2013) 256-266. DOI:10.1038/nrclinonc.2013.42 |
| [22] |
R.A. Juergens, J. Wrangle, F.P. Vendetti, et al., Cancer Discov. 1 (2011) 598-607. DOI:10.1158/2159-8290.CD-11-0214 |
| [23] |
E.A. Griffiths, S.D. Gore, Semin. Hematol. 45 (2008) 23-30. DOI:10.1053/j.seminhematol.2007.11.007 |
| [24] |
Z. Cui, X. Li, L. Li, et al., Bioorg. Med. Chem. 24 (2016) 261-269. DOI:10.1016/j.bmc.2015.12.011 |
| [25] |
W. Zhang, B. Zhang, W. Zhang, et al., Eur. J. Med. Chem. 116 (2016) 59-70. DOI:10.1016/j.ejmech.2016.03.066 |
| [26] |
C. Ding, D. Li, Y.W. Wang, et al., Chin. Chem. Lett. 28 (2017) 1220-1227. DOI:10.1016/j.cclet.2017.01.003 |
| [27] |
Z. Yuan, Q. Sun, D. Li, et al., Eur. J. Med. Chem. 134 (2017) 281-292. DOI:10.1016/j.ejmech.2017.04.017 |
| [28] |
S. Chen, Y. Wang, W. Zhou, et al., J. Med. Chem. 57 (2014) 9028-9041. DOI:10.1021/jm501134e |
| [29] |
A. Scuto, M. Kirschbaum, C. Kowolik, et al., Blood 111 (2008) 5093-5100. DOI:10.1182/blood-2007-10-117762 |
| [30] |
J. Datta, K. Ghoshal, W.A. Denny, et al., Cancer Res. 69 (2009) 4277-4285. DOI:10.1158/0008-5472.CAN-08-3669 |
| [31] |
A.K. Witkiewicz, K.E. Knudsen, A.P. Dicker, E.S. Knudsen, Cell Cycle 10 (2011) 2497-2503. DOI:10.4161/cc.10.15.16776 |
| [32] |
D. Hanahan, R.A. Weinberg, Cell 144 (2011) 646-674. DOI:10.1016/j.cell.2011.02.013 |
| [33] |
K. Ha, W. Fiskus, D.S. Choi, et al., Oncotarget 5 (2014) 5637-5650. |
| [34] |
Q. Wu, W. Xu, L. Cao, et al., J. Proteome Res. 12 (2013) 4064-4073. DOI:10.1021/pr4004079 |
| [35] |
H.G. Zhang, W.E. Grizzlet, Am. J. Pathol. 184 (2014) 28-41. DOI:10.1016/j.ajpath.2013.09.027 |
| [36] |
M.S. Sosa, C. Lopez-Haber, C. Yang, et al., Mol. Cell 40 (2010) 877-892. DOI:10.1016/j.molcel.2010.11.029 |