 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, CAS, Fuzhou 350002, China
Carboxylic acids are widely found in nature, which are a kind of nontoxic and inexpensive compounds. As a result, one of the current research interests is the transition-metal catalyzed carboxylate-directed transformation or decarboxylative conversion to form newand etero bonds [1]. Along this line, we have gained extensive knowledge in the realm of Pd-mediated decarboxylative ortho-halogenation of aromatic carboxylic acids using carboxyl as traceless directing group and Cu-promoted decarboxylative ipso-functionalization of aryl carboxylic acids [[2]], including decarboxylative methylthiolation [3a], arylation [3b], protonation [2c], cyanation and halogenation [2d–f]. In addition, carboxylic acids can also be employed to synthesize esters, it is worth to note ester scaffolds serve as value intermediates in organic synthesis and key structural units in a variety of natural products, pharmaceuticals and polymers [[3]]. As a consequence, the development of efficient methods for facile construction of organic esters molecules has attracted considerable attention in the past several decades. The traditional method toward the condensation of carboxylic acids or their derivatives with alcohols represents one of the most conventional methods to synthesize corresponding esters, although the drawbacks associated with incomplete transformation and/or generation of a large amounts of unwanted by-products [[3]]. Moreover, great progress has been made to prepare esters via catalytic coupling of carboxylic acids with various partners including aryl organometallic reagents [4], aldehydes [5], and arenes [6]. More recently, significant attention has been paid to esterification conversion via cross coupling of carboxylic acids with aryl halides alo bond activation, in this regard, McCusker and MacMillan firstly reported the coupling of carboxylic acids with aryl bromides to provide aryl esters under excited-state organometallic catalysis via direct photoexcitation [7a], whereas Itami described carboxylic acids and aryl iodides underwent a transition-metal Pd-catalyzed coupling reaction to furnish the desired aryl esters [7b].
Compared to aryl halides, dichloromethane (DCM) is one of the simplest and cheapest halogenated hydrocarbon molecules that widely used as solvent in laboratory, in addition, methylene chloride can also be employed as reagent to build newbond and etero bond. On this point, with double C-Cl activation of dichloromethane, Kozak [8a] and Zhang [8b] disclosed the insertion of methylene fragment of DCM into two equivalents of Grignard reagents or oxazolidin-2-ones partner using iron or NaH as promoter respectively. To the best of our knowledge, it is seldom used DCM as C1 synthon in the synthesis of methylene-bridged acylals [9], which are important starting material for the preparation of dienes and chiral allylic esters in organic synthesis [10]. As one part of our continuous efforts to develop efficient functionalization transformation of cheap and abundant carboxylic acids [3], herein we reveal a simple, effective, metal-free and scalable approach for the synthesis of methylene diesters via cross coupling of dichloromethane with various carboxylic acids using K2CO3 as the sole additive.
Unless otherwise noted, the reagents used for experiments were commercially available and were used as received. All reactions were performed in dried glass reaction tube equipped with a magnetic stir bar under air atmosphere. Column chromatography was performed on silica gel mesh. The yields reported are the isolated yields and the average of two runs. 1H, 13C and 19F NMR spectra of compounds were recorded at 400, 100 and 377 MHz with CDCl3 as solvent respectively. All coupling constants (J values) were reported in Hertz (Hz). HRMS were performed by Center of Analysis and Testing, Nanchang University.
The research was commenced by performing the coupling of 1- adamantanecarboxylic acid 1 (1-AdCOOH) with CH2Cl2 as the model reaction for the optimization studies, in which CH2Cl2 was used as a solvent and stoichiometric methylene source. Table 1 presented some selected results from the optimization studies, which illustrated the effects of the additive, temperature, reaction time and other reaction parameters on the transformation outcome. As our recently developed decarboxylative ipso-functionalization reactions have been performed at 140 ℃ [2a–d], initially, in the presence of 1 equiv. of Li2CO3 as additive, the model reaction between CH2Cl2 and 1-AdCOOH 1 was performed at 130 ℃ without metal mediator under an air atmosphere to provide desired methylene diadamantanecarboxylate 1p in 28% yield (Table 1, entry 1). To improve conversion of the reaction, a careful examination of different alkali carbonates was next performed, which revealed that K2CO3 gave superior yield than that of Li2CO3 and Na2CO3 under otherwise identical conditions (entries, furthermore, upon evaluating the effect of the counterion of potassium salt compound on the transformation, it is found K2CO3 was a much better choice than either K3PO4 or KOtBu (entries. Nevertheless, lower yields were obtained when employing organic base instead of K2CO3 as additive for the model reaction (entry 6). It is noteworthy that doubling of K2CO3 inputs resulted into enhanced reaction efficiency in synthetically useful levels (88%) (entry 7). To our delight, reaction temperature was an important factor to affect the conversion efficiency, since lowering temperature to 100 ℃ exerted a detrimental effect on the reaction and delivered the corresponding product 1p in 75% yield (entry 8). It is gratifying to obtain as high as 93% yield of the desired product 1p when the reaction time was reduced to 5 h (entry 9), unfortunately, both switching mixed solvent CH2Cl2/DMSO to CH2Cl2/NMP and CH2Cl2/DMF and changing reaction atmosphere to aerobic conditions led to inferior reaction conversion under the same reaction time (5 h, entries 10 and 11).
|
|
Table 1 Optimization of the reaction conditions.a |

Finally, controlled experiment showed that none of target product 1p could be detected in the absence of K2CO3, suggesting that the base is essential for this conversion (entry 12). Weighing the pros and cons, the coupling of dichloromethane with carboxylic acids using 2 equiv. of K2CO3 as the sole additive at 130 ℃ for under an air atmosphere was employed as standard conditions to synthesize methylene diesters.
Subsequently, the obtained optimality conditions (entry 9, Table 1) to achieve methylene diadamantanecarboxylate 1p encouraged us to evaluate the scope of this metal-free coupling of methylene chloride with respect to carboxylic acids, as illustrated in Table 2, the transformation was shown to be an effective strategy for a diverse array of alkyl carboxylic acids 1-7, α, β-unsaturated carboxylic acids 8-10, heteroaryl carboxylic acids 11-22 and α-oxobenzeneacetic acid 23. Besides tertiary aliphatic carboxylic acids 1 and 2 provided the desired products in good to excellent yields in this protocol, cyclohexanecarboxylic acid 3 also proceeded straightforward with DCM to give product 3p in nearly quantitative yield. Remarkablely, primary carboxylic acids including aminoacetic and oxyacetic acid 6 and 7 as well as the substrates 4 and 5 tolerated carbon-carbon double bond and aryl functional groups, which were well carried out with CH2Cl2 under the standard conditions. For the substrates α, β-unsaturated carboxylic acids, crotonic acid 8 and cinnamic acid 9 successfully furnished desired products in nearly quantitative yield, cinnamalacetic acid 10 comprised a conjugated diene unit also generated the end product, albeit with mild conversion. Gratifyingly, O-, S- and N-containing heteroaryl carboxylic acids 11-22 were proved to be suitable starting materials to afford hoped-for products. Significantly, it is no matter whether carboxyl group at the position ortho or meta to the sulfur, thiophenic acids 12 and 13 were also quantitatively transformed to corresponding products. Even though a methyl substituent presented on the position ortho to the carboxyl, the substrates 16-19 and 21 gave expected products in good to excellent yields without being affected by the steric effect. Finally, α-oxobenzeneacetic acid 23 delivered the desired product in moderate yield, and it's failed to improve conversion efficiency through increasing reaction temperature or enhancing the amount of K2CO3 additive.
|
|
Table 2 Scope of carboxylic acids in the transformation.a |
The feasibility of coupling of dichloromethane with alkyl and heteroaryl carboxylic acids inspired us to further explore the generality of other aromatic carboxylic acids with different substituents. As illustrated in Table 3, a wealth of benzoic acids were favorably carried out with CH2Cl2 under the standard conditions. It is observed that benzoic acid 32 was a viable substrate to afford desired methylene dibenzoate 32p with CH2Cl2 in quantitative yield, which was the divide between electronwithdrawing (fluoro, chloro, cyano, nitro and sulfonyl) and electron-donating (methyl, methoxy and morpholinyl) substituents on the benzoic acid substrate, thus, the substituent groups were found to be broad and well-tolerated in this protocol. Apart from the substrates 24, 26, 28 and 29, the conversion efficiency of benzoic acids bearing electron-donating groups was superior to that of benzoic acids with electron-withdrawing groups in this protocol, which could be explained as a result of enhanced electron density of aryl carboxylate through electron-donating substituent on the aromatic ring in the presence of K2CO3. Moreover, steric hindrance did not significantly affect the efficiency of the reaction, compared with their isomers, benzoic acids (27, 29, 31, 37 and 39) with ortho group gave target products in good to excellent yields. No matter where the nitro or methoxy substituent was in the carboxyl group, nitrobenzoic acids and methoxybenzoic acids were effective substrates to conveniently provided corresponding products in good yields. Notably, the substituent of halo-moiety in substrate 25 survived well in the reaction, which offered the possibility for late-stage functionalization via the activation of carbon-halogen bond.
|
|
Table 3 Scope of benzoic acids in the transformation.a |

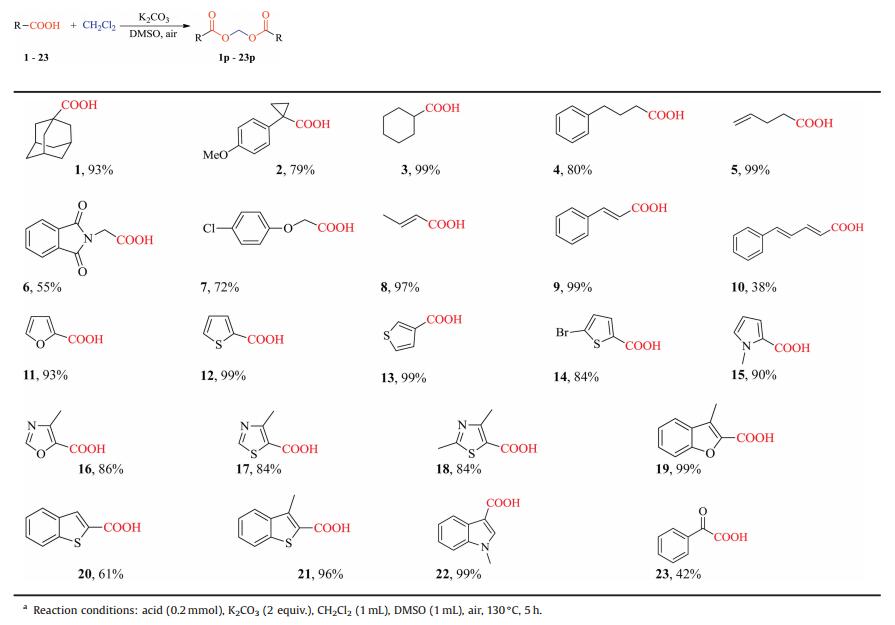
To prove the effectiveness and practicality of this method, a 40- fold scale-up of the metal-free synthesis was performed under the optimized conditions. As shown in Scheme 1, employing 8 mmol of carboxylic acid as starting material with K2CO3 (2 equiv.) in DMSO under an air atmosphere, as a result, no matter what type of carboxylic acid successfully reached nearly the same efficiency as the reaction conducted on a 0.2 mmol scale, which declared a great potential of the method on large-scale synthesis.

|
Download:
|
| Scheme 1. Gram-scale synthesis of products 1p, 9p, 11p and 32p. | |
{kind=link}
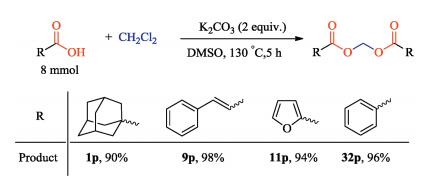
Basing on the above results, though the proposal of a detailed mechanism should be awaited further investigation, a plausible mechanism for this transformation was hypothesized in Scheme 2. Initially, the carboxylic acid substrate was easily transformed to potassium carboxylate salt in the presence of stoichiometric amounts of K2CO3, subsequently, the carboxylate anion's negatively charged oxygen nucleophilic attacked the considerably electrophilic methylene chloride's carbon. As a result, the final methylene diester was generated with the liberation of KCl product. Due in part to the decreased electron density of carboxylate by induced effect of heteroatom, the transformation yields with using benzoic acids were generally better than that employing heteroaryl carboxylic acids. Since K2CO3 was unable to regenerate during the process, that is why stoichiometric amounts of additive were needed in the reaction.

|
Download:
|
| Scheme 2. Proposed mechanism for the transformation. | |
{kind=link}
In summary, we have established a simple and practical method for the metal-free coupling reaction between easily available CH2Cl2 and carboxylic acids with K2CO3 as the sole additive. The transformation demonstrated an efficient protocol to synthesize methylene diesters in fair to excellent results with several distinguishing features: extensive substrate scope including readily available carboxylic acids with good functional group tolerance; a simple reaction system to efficiently offer corresponding esters products with cheap K2CO3 as the sole additive under metal-free conditions. Further synthetic application of the protocol is currently in progress.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (NSFC, Nos. 21761021 and 21571094), Natural Science Foundation of Jiangxi Province (No. 20171BAB203002), Sci & Tech Project of Education Department of Jiangxi Province (No. 60007) is gratefully acknowledged.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.053.
| [1] |
(a) C. Sambiagio, D. Schönbauer, R. Blieck, et al., Chem. Soc. Rev. 47(2018) 6603-6743; (b) Y. Wei, P. Hu, M. Zhang, W. Su, Chem. Rev. 117(2017) 8864-8907; (c) M. Font, J.M. Quibell, G.J.P. Perry, I. Larrosa, Chem. Commun. 53(2017) 5584-5597; (d) M.P. Drapeau, L.J. Gooßen, Chem.-Eur. J. 22(2016) 18654-18677; (e) M.X. Bi, P. Qian, Y.K. Wang, Z.G. Zha, Z.Y. Wang, Chin. Chem. Lett. 28(2017) 1159-1162. |
| [2] |
(a) Z. Fu, Z. Li, Q. Xiong, H. Cai, Eur. J. Org. Chem (2014) 7798-7802; (b) Z. Fu, Z. Li, Q. Xiong, H. Cai, RSC Adv. 5(2015) 52101-52104; (c) Z. Li, Z. Fu, H. Zhang, J. Long, Y. Song, H. Cai, New J. Chem. 40(2016) 3014-3018; (d) Z. Fu, Z. Li, Y. Song, et al., J. Org. Chem. 81(2016) 2794-2803; (e) Z. Fu, L. Jiang, Q. Zuo, et al., Org. Biomol. Chem. 16(2018) 5416-5421; (f) Z. Fu, Y. Jiang, L. Jiang, et al., Tetrahedron Lett. 59(2018) 4458-4461. |
| [3] |
(a) R.C. Larock, Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, New York, 1999; (b) J. Otera, Esterification, Wiley-VCH, Weinheim, 2003. |
| [4] |
(a) F. Luo, C. Pan, P. Qian, J. Cheng, Synthesis (2010) 2005-2010; (b) L. Zhang, G. Zhang, M. Zhang, J. Cheng, J. Org. Chem. 75(2010) 7472-7474; (c) J.J. Dai, J.H. Liu, D.F. Luo, L. Liu, Chem. Commun. 47(2011) 677-679; (d) W.H. Dong, D.D. Wu, J.M. Luo, et al., J. Catal. 349(2017) 218-225. |
| [5] |
(a) H. Toledo, E. Pisarevsky, A. Abramovich, A.M. Szpilman, Chem. Commun. 49(2013) 4367-4369; (b) D. Saberi, F. Shojaeyan, K. Niknam, Tetrahedron Lett. 57(2016) 566-569. |
| [6] |
(a) S. Moghimi, M. Mahdavi, A. Shafiee, A. Foroumadi, Eur. J. Org. Chem. (2016) 3282-3299; (b) I.B. Krylov, V.A. Vil', A.O. Terent'ev, Beilstein J. Org. Chem. 11(2015) 92-146; (c) G. Majji, S.K. Rout, S. Rajamanickam, S. Guina, B.K. Patel, Org. Biomol. Chem. 14(2016) 8178-8211; (d) J.P. Zou, D.D. Wu, J. Luo, et al., ACS Catal. 6(2016) 6861-6867. |
| [7] |
(a) E.R. Welin, C. Le, D.M. Ariasrotondo, J.K. Mccusker, D.W.C. Macmillan, Science 355(2017) 380-385; (b) H. Kitano, H. Ito, K. Itami, Org. Lett. 20(2018) 2428-2432. |
| [8] |
(a) X. Qian, C.M. Kozak, Synlett 2011(2011) 852-856; (b) Q. Liu, Y. Zhang, Z. Zhang, et al., RSC Adv. 4(2014) 25933-25939. |
| [9] |
(a) F. Lin, Q. Feng, X. Cui, Q. Song, RSC Adv. 3(2013) 20246-20253; (b) Y.H. Ma, G. Wu, N. Jiang, et al., Chin. Chem. Lett. 26(2015) 81-84. |
| [10] |
Y. Ishino, M. Mihara, Tetrahedron Lett. 45 (2004) 3503-3506. DOI:10.1016/j.tetlet.2004.02.159 |