 2019, Vol. 30
2019, Vol. 30 


b Institute of Theoretical and Computational Chemistry, Shaanxi Key Laboratory of Catalysis, School of Chemical & Environment Sciences, Shaanxi University of Technology, Hanzhong 723000, China;
c State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China;
d Hefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei 230026, China
Metal/metal oxide composites have a wide range of applications, such as microelectronics [1], gas sensors [2], photovoltaics [3-8] and catalysis [9-11]. Among the metal/metal oxide composites in catalysis, nickel-based catalysts have been widely used in broad applications, e.g., hydrogenation, hydrotreatment and hydrogenolysis of hydrocarbons, steam reforming of hydrocarbons, and methanation [12-16], due to its low cost and high catalytic activity. However, nickel-based catalysts have the disadvantage of deactivation due to the aggregation of nickel [17, 18]. The growth of Ni atoms/clusters into large particles is inevitable in the catalytic reaction, which finally leads to the deactivation of the catalysts greatly affects its performance. Theoretical investigations have been carried out on the mechanisms of the growth and aggregation of Ni atoms on γ-Al2O3 surfaces [19]. It found that Ni growth into larger particle is thermodynamically favorable on the γ-Al2O3 support and its surface hydroxyls can inhibit the aggregation of clusters. It is also found that Cu growth into larger particle is thermodynamically favorable [20]. The surface hydroxyls formed by adsorbed water on γ-Al2O3 surfaces could inhibit the growth of Cu.
In catalysis, Ni particles could be supported on different individual metal oxide supports, such as γ-Al2O3 [21, 22], SiO2 [23], La2O3 [24], TiO2 [25], CeO2 [26, 27]. However, some composite metal oxides having a fixed chemical formula and high thermal stability, such as perovskite (ABO3) and spinel (AB2O4) [28-31]. Aimed at the development of catalysts with stronger coking resistance and higher activity, Gao et al. [32] found that the CuAl2O4 spinel catalysts perform high activity and stability for methanol steam reforming (MSR) since the Cu atoms in the CuAl2O4 spinel surface are single active sites, and the Cu atoms in the spinel could be gradually released in the reduction atmosphere, which leading to the increased activity in the initio stage of the reaction. The problem is that the CuAl2O4 spinel catalysts also goes to deactivation during long catalysis run. Experiments show that the stability of Cu-Al spinel can be greatly improved by adding a small amount of Ni, Mg, B additives, and the effect of Ni is the best [33]. However, the mechanisms for the interaction of Ni atoms with the CuAl2O4 spinel surface, which is fundamental for understanding the stability of the catalysts, is unclear.
In the present work, spin-polarized dispersion corrected density function theory (DFT) calculations were performed for the investigation of the interaction of Ni atoms with CuAl2O4 spinel surface. Since the CuAl2O4 spinel catalysts usually work under reduction atmosphere and the surface oxygen atoms maybe pull out by hydrogen to produce water and oxygen vacancies in the oxide surface, the adsorption of Ni atom on the O-defect sites of CuAl2O4 spinel surface is also investigated.
All calculations were performed with the PBE-D method with plane wave basis set as implemented in the Vienna ab initio Simulation Package (VASP) [34, 35]. The D3 method of Grimme was chosen to calculate the dispersive interactions for the DFT optimized structures [36]. The exchange and correlation energies were calculated by the generalized gradient approximation (GGA) formulation with the PBE functional [37]. The Kohn-Sham oneelectron states were extended in accordance with plane-wave basis sets with the cut off energy of 400 eV. The projector augmented wave (PAW) method was applied to describe the electron-ion interactions [38, 39]. The Brillouin zone was sampled with 5 × 5×5 and 3 × 3×1 k-points meshes for the unit cell and surface slabs, respectively, generated by the Monkhorst-Pack algorithm. The convergence criteria were set 1.0 ×10–5 eV for the SCF energy, 1 ×10–4 eV and 0.03 eV/Å for the total energy and the atomic forces, respectively.
The adsorption energies (Eads) for the adsorption of Nin (n = 1–4) in the surface were calculated by:

|
(1) |
Where E(Nin/slab), E(Nin) and E(slab) are the total energies of the spinel surface slab with adsorbed Ni atoms/clusters, isolated the gas phase Ni atoms/clusters and the spinel surfaces, respectively.
The growth energy is defined as the energy for the formation of the n-atom cluster/slab from the (n-1)-atom cluster/slab and one Ni atom/slab:
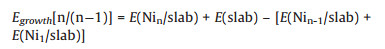
|
(2) |
The aggregation energy for Ni clusters on the surface is defined as:
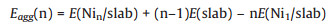
|
(3) |
The average binding energy of isolated Nin (n = 2–4) clusters, Ebind(Nin) is calculated as follows:
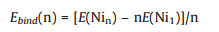
|
(4) |
It was reported that the CuAl2O4 is of Fd-3 m space group, as show in Fig. 1a, Cu2+ cations are in tetra-coordination and Al3+ cations in the octa-coordination [40]. All O2- anions are in tetra-coordination. The unit cell of spinel contains Cu8Al16O32, with the optimized lattice parameter of 8.1669 Å, which is in good agreement with the experiments results of 8.0778–8.153 Å [41, 42]. Our calculated Cu-O and Al-O bond distances are 198 and 193 pm, respectively. The surface Cu-O distance is larger than that of Cu2O [43].

|
Download:
|
| Fig. 1. CuAl2O4 spinel surface structures. (a) CuAl2O4 spinel unit cell; (b) (100) surface; (c) (110) surface. O, Al and Cu atoms are in red, gray and blue, respectively. The exposed top layer atoms are indexed with numbers and the coordination numbers are shown in subscript. | |
Figs. 1b and c show the side and top views of the CuAl2O4 spinel (100) and (110) surfaces. Both the (100) and (110) surface slabs contain the unit of Cu16Al32O64. The bottom half (below the dashed line, Cu8Al16O32) of the slabs are fixed in their bulk position and the top half (on the dashed line, Cu8Al16O32) of the slab are fully relaxed for all optimization calculations.
The top layer of the perfect (100) surface exposes Cu(2, 4)2c as equivalent di-coordinated Cu atoms, whereas Cu(1, 3)4c are tetracoordinated in the slightly lower than the top layer atoms. O (3, 5, 6, 8)3c are equivalent tri-coordinated and O(1, 2, 4, 7)4c are equivalent tetra-coordinated O atoms. All Al(1, 2, 3, 4)5c atoms in the surfaces are equivalent penta-coordinated. The perfect (110) surface exposes equivalent Cu(1, 2, 3, 4)3c in tri-coordination, equivalent O(1, 2, 3, 4, 5, 6, 7, 8)3c in tri-coordination and equivalent Al(1, 2, 3, 4)4c in tetra-coordination of the top layer, whereas, equivalent O(9, 10, 11, 12)4c are tetra-coordination in the sublayer.
It is expected that the equivalent surfaces atoms, e.g. Cu(2)2c and Cu(4)2c; Cu(1)4c and Cu(3)4c; O(3)3c O(5)3c O(6)3c and O(8)3c; O (1)4c, O(2)4c, O(4)4c and O(7)4c; Al(1)5c, Al(2)5c, Al(3)5c and Al(4)5c atoms in (100) surface, as well as the Cu(1)3c, Cu(2)3c, Cu(3)3c and Cu(4)3c; O(1)3c, O(2)3c, O(3)3c, O(4)3c, O(5)3c, O(6)3c, O(7)3c and O (8)3c; Al(1)4c, Al(2)4c, Al(3)4c and Al(4)4c, should have the same chemical properties for Ni atom adsorption.
To get the defective surface with oxygen defects, we tried to remove one of the surface O atoms from both (100) and (110) surfaces. It is found that O(3)3c atoms are the most favored one to be removed from both the (100) and (110) surface. The formation of one oxygen-defective surface and half gas phase O2 molecule from the perfect (100) and (110) surfaces are thermodynamically endothermic by 1.61 and 2.64 eV, respectively. It indicates that it is more easily to create the O defects in the (100) surface than in the (110) surface.
After the removal of the surface O(3)3c atoms, both (100) and (110) surface are distorted. As shown in Fig. 1b, the O-defect (100) O(6)2c and O(4)3c become di- and tri-coordinated, Al(1)4c and Al (2)4c become tetra-coordinated, Cu(1)3c and Cu(3)3c are tricoordinated, respectively. The O-defect (110) surface distorted even more serious than the (100) surface. The O-defect (110) surface shows that Cu(4)2c become di-coordinated, and the surface O(1, 2, 4, 5, 6, 7, 8) and O(9, 10, 11, 12) keeps their tri- and tetra-coordination, respectively. However, it is found that the Al(3)4c atom moves to sublayer, and accordingly one of the sublayer Al moves to left.
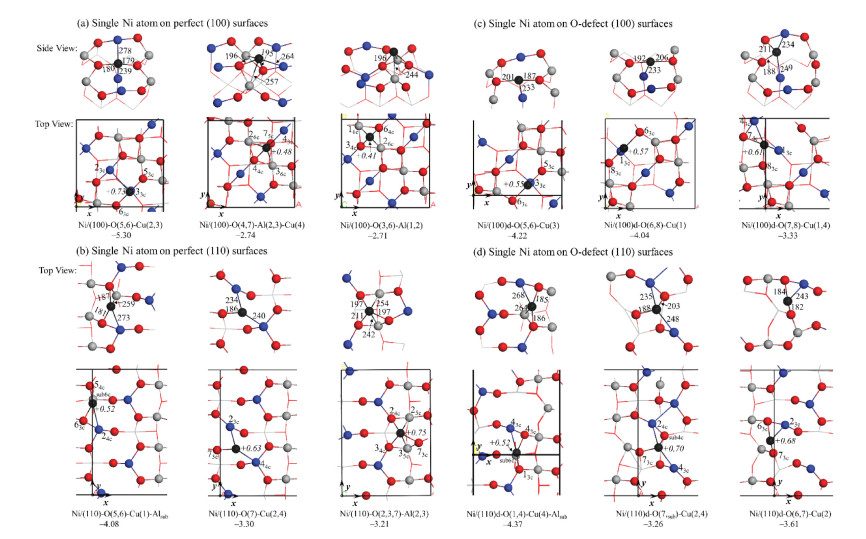
The gas phase molecules are calculated using an a = b = c = 15 Å cell. The structures of single Ni atom adsorbed the perfect CuAl2O4 spinel (100) surface are presented in Fig. 2a The results show that Ni atom bonds with the O(5, 6) and Cu(2, 3), as shown in Ni/(100)-O (5, 6)-Cu(2, 3), is the most stable adsorption structure, which releases the adsorption energy of -5.30 eV. Bader charge analysis shows that the adsorbed Ni atom in Ni/(100)-O(5, 6)-Cu(2, 3) is +0.73 e, with Ni-O distances of 179 and 180 pm, Ni-Cu distances of 239 and 278 pm, respectively.

|
Download:
|
| Fig. 2. Optimized adsorption configurations and the adsorption energies (eV) for single Ni atom on perfect and the O-defect CuAl2O4 spinel (100) and (110) surfaces. O, Al, Ni and Cu atoms are shown in red, gray and black and blue balls, respectively. The bond distances and Bader charges are in pm and e. | |
For Ni adsorbed on other sites of the (100) surface, e.g., in Ni/ (100)-O(4, 7)-Al(2, 3)-Cu(4) and Ni/(100)-O(3, 6)-Al(1, 2), the adsorption energies are -2.74 and -2.71 eV, respectively, which are about 2.60 eV less stable than in Ni/(100)-O(5, 6)-Cu(2, 3). It indicates that the Ni atom on the spinel (100) surface prefers to be adsorbed on the Ni/(100)-O(5, 6)-Cu(2, 3) site.
Three local minima structures were found for single Ni atom adsorbs on the perfect CuAl2O4 (110) surface. As shown in Fig. 2b, in themost stable configuration, single Ni located atNi/(110)-O(5, 6)-Cu (1)-Alsub with adsorption energy of -4.08 eV. In this structure, the Ni (+0.52e)bonds with the O(5, 6)and Cu(1), as well as sublayer Al, with the Ni-O distances of 181, 187 pm, Ni-Cu distances of 273 pm and Ni- Al distances of 259 pm, respectively. The adsorption energy of single Ni at Ni/(110)-O(7) -Cu(2, 4) is -3.30 eV, which is less stable than in Ni/(110)-O(5, 6)-Cu(1)-Alsub. The Ni (+0.63 e) connect to O(7) and Cu(2, 4), with the Ni-O distances of 186 pm, Ni-Cu distances of 234, 240 pm, respectively. In Ni/(110)-O(2, 3, 7)-Al(2, 3), the Ni is +0.75 e with Ni-O and Ni-Al distances of 197, 211 and 242, 254 pm, respectively, and the adsorption energy of -3.21 eV. It should be mentioned that though obvious charge transfer from the adsorbed Ni atom to the surface are confirmed by the Bader charge analysis. No correlation was found between the charge transfer and calculated adsorption energy.
Figs. 2c and d show the structures and adsorption energies for single Ni atom on the O-defect CuAl2O4 spinel (100) and (110) surfaces. It shows that Ni/(100)d-O(5, 6)-Cu(3) is the most stable adsorption site for Ni adsorbed on the O-defect (100) surface. In this structure, the Ni is +0.55 e, with the bond distance of 187 and 201 pm for Ni-O, and 233 pm for Ni-Cu, respectively. It should be noted that the adsorption energy for Ni/(100)d-O(5, 6)-Cu(3) is -4.22 eV, which is 1.08 eV less stable than in Ni/(100)-O(5, 6)-Cu (2, 3). It indicates that the Ni adsorbed on the O-defect (100) surface is less stable than on the perfect surface. In Ni/(100)d-O(6, 8)-Cu(1), the Ni atom (+0.57 e) bonds with the O(6, 8) and Cu(1) atoms, with the Ni-O distances of 192, 206 pm and Ni-Cu distances of 233 pm. The adsorption energy for single Ni located in Ni/(100)d-O(6, 8)-Cu (1) is -4.04 eV, which is close to Ni/(100)d-O(5, 6)-Cu(3). The adsorption energy of single Ni at Ni/(100)d-O(7, 8)-Cu(1, 4) is -3.33 eV, which is weaker than Ni adsorbed on Ni/(100)d-O(5, 6)- Cu(3) and Ni/(100)d-O(6, 8)-Cu(1).
As shown in Fig. 2d, the most stable adsorption site for single Ni atom on O-defect CuAl2O4 (110) surface is Ni/(110)d-O(1, 4)-Cu(4)- Alsub. The adsorption energy is -4.37 eV, which is 0.29 eV more stable than Ni adsorbed on Ni/(110)-O(5, 6)-Cu(1)-Alsub. The Ni atom (+0.52 e) bonds with the O(1, 4), Cu(4), and sublayer Al atom. With the Ni-O, Ni-Cu, Ni-Al distances of 185, 186, 268, 264 pm, respectively. In Ni/(110)d-O(7, sub)-Cu(2, 4), the adsorption energy is -3.26 eV, with the Ni atom (+0.70 e) bonds to the O(7), Cu(2, 4) and sublayer O atom. The Ni-O distances are 188 and 203 pm vs. Ni-Cu distances of 235 and 248 pm, respectively. The adsorption energy of Ni (+0.68 e) in Ni/(110)d-O(6, 7)-Cu(2) is -3.61 eV, with the Ni-O distance of 182 and 184 pm, and Ni-Cu distance of 243 pm.
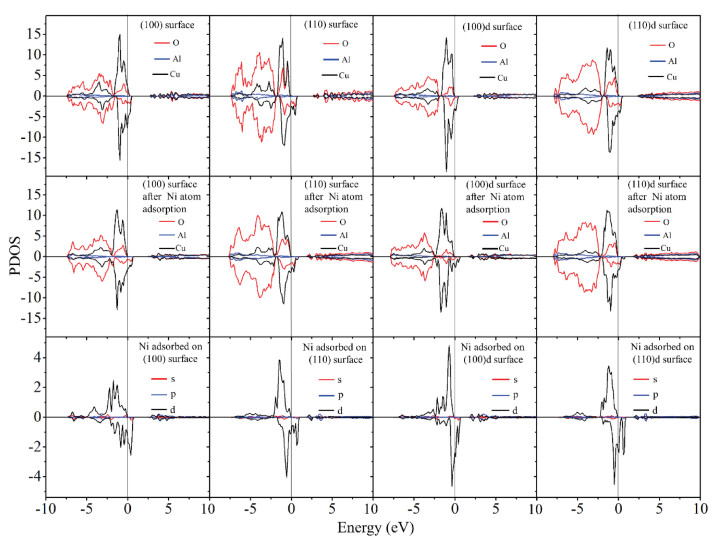
In order to investigate the electronic structurefor the adsorbed Ni and spinel surface, PDOS for the adsorbed Ni atom, top layer Cu, Al, and O atoms of the slab for Ni adsorption on perfect and O-defect CuAl2O4 (100) and (110) surfaces are shown in Fig. 3. It shows that after the removal of one surface O atom to produce the oxygen defects in both of the (100) and (110) surface, the d orbital of the surface Cu atom moves to lower energy level and become more occupied than the perfect surface. This is because the Cu atom in the defect surface are less oxidized than in the perfect surface.

|
Download:
|
| Fig. 3. PDOS for the adsorbed Ni atom, top layer Cu, Al, and O atoms of the slab for Ni adsorption on perfect and O-defect CuAl2O4 (100) and (110) surfaces. See the structure in Fig. 2. Ni/(100)-O(5, 6)-Cu(2, 3), Cu/(110)-O(5, 6)-Cu(1)-Alsub, Ni/(100)d-O(5, 6)-Cu(3) and Ni/(110)d-O(1, 4)-Cu(4)-Alsub. | |
It is well known that the isolated Ni atom has a shell electronic structure of 3d84s2, showing its 3d orbitals are electron unsaturated. After the adsorption of Ni on the perfect and O-defect (100) and (110) surface, the unoccupied orbitals of the surface O and Cu atoms near the fermi level became occupied. In accordance, the s orbitals and parts of the d orbitals of the Ni atoms deplete accompanied by the broaden of the d orbitals the adsorption of Ni atom on the surfaces. It clearly demonstrates the charge transfer from the Ni atom to the CuAl2O4 spinel surfaces.
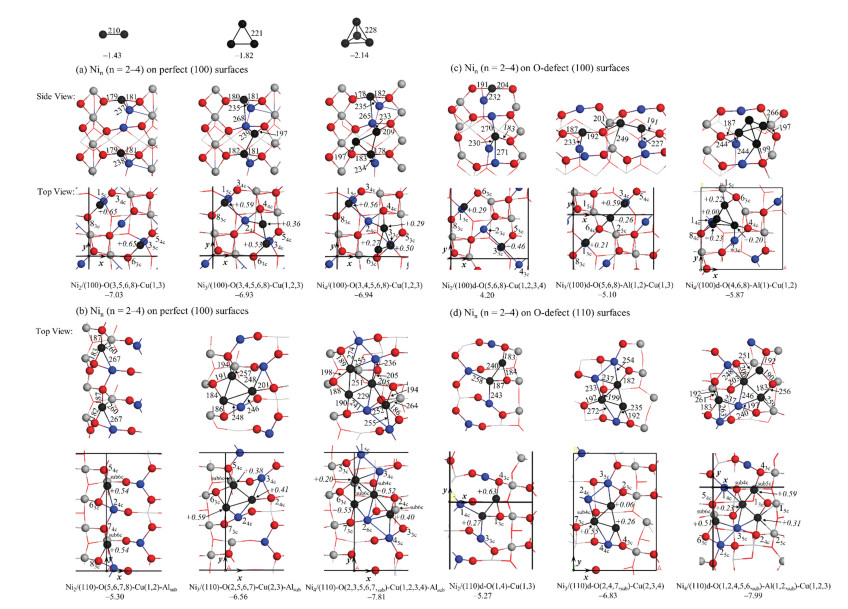
The optimized the most stable constructions for gas phase Nin (n = 2–4) clusters and their binding energy are shown in Fig. 4. The binding energies for Ni2, Ni3 and Ni4 are -1.43, -1.82 and -2.14 eV, respectively, with the average bond distances of 210, 221 and 228 pm. It indicates that the Ni-Ni interaction become stronger as the Ni cluster growth larger. Based on the most stable adsorption site for single Ni atom adsorption, we further tried to put the second, third and fourth Ni atoms on the perfect and O-defect CuAl2O4 (100) and (110) surfaces.

|
Download:
|
| Fig. 4. Gas phase Nin (n = 2–4) clusters and their adsorption on the perfect CuAl2O4 spinel surfaces. O, Al, Ni and Cu atoms are shown in red, gray and black and blue balls, respectively. The bond distances, Bader charges and adsorption energies (binding energies for Nin) are in pm, e and eV, respectively. | |
The results for Nin (n = 2-4) adsorption on the perfect CuAl2O4 (100) and (110) surface are shown in Figs. 4a and b. For Ni2 adsorption on the perfect CuAl2O4 (100) surface, the most stable adsorption site is Ni2/(100)-O(3, 5, 6, 8)-Cu(1, 3) with the adsorption energy is -7.03 eV. Since the interaction of single Ni atom adsorbed on CuAl2O4 (100) surface (-5.30 eV) is stronger than the Ni-Ni interaction (-2.86 eV) in Ni2, Ni2 on the perfect spinel CuAl2O4 (100) surface forms dissociative adsorption. The two Ni atoms of Ni2 dissociates and bond to O(3, 8)-Cu(1) and O(5, 6)-Cu(3), respectively, with the two Ni-O distance of 179 and 181 pm and Ni-Cu distance around 237 pm. For Ni3 clusters, the most stable adsorption site is Ni3/(100)-O(3, 4, 5, 6, 8)-Cu(1, 2, 3) with the adsorption energy of -6.93 eV, which is smaller than the adsorption for Ni2. In this configuration, the third Ni atom interact directly with pre-adsorbed Ni, O(4) and Cu(2) form the adsorbed Ni2 with another Ni atom. The bond distance of Ni-Ni is 228 pm, which is 7 pm elongated compared to that in gas phase Ni3. For Ni4 clusters, the forth Ni atom bond with the pre-adsorbed Ni2, which forms a triangle, and the average bond length of Ni-Ni is 230 pm which is larger than that of Ni4 clusters (228 pm) in gas phase. The adsorption energy of in Ni4/(100)-O(3, 4, 5, 6, 8)-Cu(1, 2, 3) is -6.94 eV, which is very close to Ni3 adsorption.
As shown in Fig. 4b, Ni2 adsorbs on the perfect spinel (110) surface also dissociates due to the strong interaction between nickel and CuAl2O4 spinel surface. The adsorption energy for Ni2/ (110)-O(5, 6, 7, 8)-Cu(1, 2)-Alsub is -5.30 eV, which is 1.73 eV less stable than in Ni2/(100)-O(3, 5, 6, 8)-Cu(1, 3). In Ni2/(110)-O(5, 6, 7, 8)- Cu(1, 2)-Alsub, the two separated Ni atoms bonds to O(5, 6)-Cu(2) and sublayer Al atoms, as well as O(7, 8)-Cu(1) and sublayer Al atom with the two Ni-O distance of 182, 183 and 187 pm, Ni-Cu distance of 267 pm and Ni-Alsub distance of 260 pm. The adsorption energy for Ni3/(110)-O(2, 5, 6, 7)-Cu(2, 3)-Alsub is -6.56 eV, which is 1.26 eV larger than Ni2 adsorbed on the CuAl2O4 (110) surface. In this structure, the adsorbed Ni3 forms a triangle structure which is consistent with the most stable structure of gas phase Ni3. The adsorption energy of Ni4 in the (110) surface is -7.81 eV, which is 1.25 eV larger than that of Ni3 adsorption in the CuAl2O4 (110) surface and Ni4 adsorption in CuAl2O4 (100) surface. Ni4 and surface Cu atoms in the surface form belt laying in Ni4/ (110)-O(2, 3, 5, 6, 7, sub)-Cu(1, 2, 3, 4)-Alsub, which is different from the gas phase tetrahedron structures of Ni4. The average Ni-Ni bond distance for Ni4 adsorbed on CuAl2O4 (110) surface is 246 pm that larger than Ni4 in gas phase. It should be mentioned that the adsorption energy of nickel clusters adsorbed on the perfect surface of copper-aluminum spinel is larger than that of copper clusters [44].
The Bader charge analysis show that the single Ni atom, Ni2, Ni3 and Ni4 in the CuAl2O4 spinel (100) surface are oxidized to +0.73, +1.30, +1.48 and +1.62 e, vs. +0.52, +1.08, +1.38 and +1.67 e in the (110) surface. It indicates that the (100) surface of CuAl2O4 spinel has stronger ability than the (110) surface to get electrons from the supported metal. The total charge transfer from the metal to the surface became larger as the Ni cluster became larger. However, the average charge transfer from per Ni atom to the spinel surface (+0.73, +0.65, +0.49 and +0.41 e in the (100) surface, vs. +0.52, +0.54, +0.46 and +0.42 e in the (110) surface) became smaller.
As shown in Fig. 4c, the most stable adsorption structure for Ni2 adsorption on the O-defect CuAl2O4 (100) surface is Ni2/(100)d-O (5, 6, 8)-Cu(1, 2, 3, 4), in which the two Ni atoms also dissociates with the adsorption energy of -4.20 eV, vs. -7.03 for Ni2 adsorption on the perfect (100) surface. It should be noted that the average Ni-O bond distance (193 pm) in Ni2/(100)d-O(5, 6, 8)-Cu(1, 2, 3, 4) is longer than Ni2 adsorption in the perfect spinel (100) surface (180 pm). This is probably because that the O-defect surface has weaker ability to get electrons from the Ni atoms, which leads to the weaker interaction between the Ni atoms with the surface, compared with the perfect surface. The adsorption energy of Ni3 clusters in Ni3/(100)d-O(5, 6, 8)-Al-(1, 2)-Cu(1, 3) is -5.10 eV, which is 1.83 eV smaller than the adsorption energy of Ni3 on the perfect CuAl2O4 (100) surface. In this configuration, the third Ni prefers to bind to one of the pre-adsorbed Ni atom form a Ni2 (Ni-Ni of 228 pm) in the surface, with another Ni atom separately bonds to O (6, 8) and Cu(1). For Ni4 clusters, it was found that 3D tetrahedron Ni4 adsorbed on O-defect CuAl2O4 (100) is most stable with the adsorption energy of -5.87 eV, which is consistent with the most stable structure of Ni4 in the gas phase.
For Nin (n = 2–4) clusters adsorption on the O-defect CuAl2O4 (100), the adsorption energy increases as the Ni clusters became larger. However, the adsorption energy of Nin (n = 2–4) clusters in the O-defect CuAl2O4 (100) is smaller than on the perfect CuAl2O4 (100) surface. This is because the charger transfer from the metal to the surface contributes to the adsorption energy, and the O-defect CuAl2O4 (100) surface show weaker ability to get electrons than the perfect CuAl2O4 (100) surface.
As shown in Fig. 4d, the most stable adsorption site for Ni2 in the O-defect CuAl2O4 (110) surface is Ni2/(110)d-O(1, 4)-Cu(1, 3). It is interesting that the Ni2 does not dissociate in this structure with the Ni-Ni bond of is 240 pm, which much longer than it in gas phase Ni2. The adsorption energy for Ni2 in Ni2/(110)d-O(1, 4)-Cu (1, 3) is -5.27 eV, which is almost the same as in the perfect CuAl2O4 (110) surface. The adsorption energy for Ni3/(110)d-O (2, 4, 7, sub)-Cu(2, 3, 4) is -6.83 eV, in which the Ni3 forms triangle. This structure can also be interpreted as the Ni3 triangle connects one Cu2 and another Cu atom in the surface and form two rhombs (Cu2Ni2 and CuNi3) share one Ni-Ni edge. For Ni4 clusters, the most stable adsorption configuration is Ni4/(110)d-O(1, 2, 4, 5, 6, sub)-Al(1, 2, sub)-Cu(1, 2, 3) with adsorption energy of -7.99 eV, with the average Ni-Ni bond distance of is 239 pm. It is also found that the adsorption energy for Nin (n = 2–4) on O-defect CuAl2O4 (110) surface increase as the Ni cluster became larger.
Table 1 shows the growth energies and aggregation energies for Nin clusters on perfect and O-defect CuAl2O4 surfaces. The aggregation energies per Ni atom of Ni cluster in the gas phase is negative. It reveals that the Ni atoms in gas phase prefer to aggregate into large particles. On the perfect CuAl2O4 (100) surface the positive aggregation energies indicate the aggregation of Ni atoms are thermodynamically unfavored. However, the aggregation energies for Ni on the O-defect (100) and (110) surfaces, as well as the perfect (110) surface is close to 0 eV, indicates the aggregation of Ni is around balance.
|
|
Table 1 Growth energies (eV), Egrowth (n/n-1), and aggregation energies (eV) per Ni atom, Eagg(n)/n, for Nin clusters (n = 1–4) in the gas phase, on the perfect and O-defect CuAl2O4(100), (110) surfaces. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The growth energy of Nin (n = 2, 3, 4) clusters on the CuAl2O4 (100) surface is obviously positive, which indicates the single Ni atom, Ni2 and Ni3 have no ability to get one more Ni atom to growth into Ni2, Ni3, and Ni4 on the CuAl2O4 (100) surface. Similar cases also found for the on the O-defect (100) and (110) surfaces. This result can also be interpreted as the Ni2, Ni3 and Ni4 clusters are preferred to give out one Ni atom to become Ni atom, Ni2 and Ni3 in the surface. For Ni clusters in gas phase, the growth of Ni cluster is always thermodynamically favorable. On the perfect CuAl2O4 (110) surface, the growth of Ni cluster is around the reaction balance.
Since the γ-Al2O3 is usually widely used as supports for Ni catalysts, we also calculated the aggregation energies per Ni atom of Nin (n = 2–4) on γ-Al2O3 (110) surface [13], e.g., -1.00, -1.00, -1.07 eV for aggregation and -2.02, -0.98, -1.28 eV for growth. It indicates that Ni clusters on γ-Al2O3 (110) is thermodynamically favored to become large particles. The general growth and aggregation ability for Nin clusters follow the order: gas phase > γ-Al2O3 (110) > CuAl2O4 (110) > CuAl2O4 (100). It indicates that the CuAl2O4 spinel (100) surface is the most favorable to acquire the highly dispersed supported Ni clusters.
The adsorption of Nin (n = 1–4) on the perfect and O-defect CuAl2O4 (100) and (110) surfaces is investigated using periodic density functional theory. The computational results show that for single Ni atom on the perfect spinel (100) surface, the adsorption energy is -5.30 eV, much larger than on other surfaces. The adsorption of Nin (n = 1–4) absorbed on the O-defect CuAl2O4 (100) surface is less stable than on the perfect CuAl2O4 (100) surface. However, the adsorption energy for Nin (n = 1–4) on the O-defect CuAl2O4 (110) surface is close to on the perfect CuAl2O4 (110) surface. Bader charge and partial density of states (PDOS) analysis revel that the adsorption of Ni on the CuAl2O4 spinel surface is accompanied by charge transfer.
As indicated by the calculated the growth energies and aggregations energies, the general growth and aggregation ability for Nin clusters follow the order: gas phase > γ-Al2O3 (110) > CuAl2O4 (110) > CuAl2O4 (100), suggesting that the presence of spinel CuAl2O4 surfaces can prevent the growth of Ni into large particles. Furthermore, we found that the interaction of Ni on spinel CuAl2O4 surface is much stronger than the γ-Al2O3 (110) surface. This result can give reasonable explanations for the experimental phenomenon that Ni supported on the CuAl2O4 spinel performs much better stability than on the γ-Al2O3.
AcknowledgmentsThis work has been supported by the National Natural Science Foundation of China (Nos. 21763018, 21673270, 21503254 and 21875096) and the Natural Science Foundation of Jiangxi Province, China (Nos. 20181BAB203016, 20181BCD40004). Dr. Xiaohu Yu also thanks the team of syngas catalytic conversion of Shaanxi University of Technology.
| [1] |
C.T. Campbell, Surf. Sci. Rep. 27 (1996) 1-111. |
| [2] |
W.T. Koo, S.J. Choi, S.J. Kim, et al., J. Am. Chem. Soc. 138 (2016) 13431. DOI:10.1021/jacs.6b09167 |
| [3] |
K. Mondal, A. Sharma, RSC Adv. 6 (2016) 83589-83612. DOI:10.1039/C6RA18102C |
| [4] |
B. Zhu, S. Wageh, A.A. Al-Ghamdi, et al., Catal. Today (2018). DOI:10.1016/j.cattod.2018.09.038 |
| [5] |
T. Tong, B. Zhu, C. Jiang, B. Cheng, J. Yu, Appl. Surf. Sci. 433 (2018) 1175-1183. DOI:10.1016/j.apsusc.2017.10.120 |
| [6] |
B. Zhu, L. Zhang, B. Cheng, J. Yu, Appl. Catal. B-Environ. 224 (2018) 983-999. DOI:10.1016/j.apcatb.2017.11.025 |
| [7] |
B. Zhu, L. Zhang, D. Xu, B. Cheng, J. Yu, J. CO2. Util. 21 (2017) 327-335. DOI:10.1016/j.jcou.2017.07.021 |
| [8] |
S. Cao, H. Li, T. Tong, et al., Adv. Funct. Mater. 28 (2018) 1802169. DOI:10.1002/adfm.v28.32 |
| [9] |
Q. Yao, Z.H. Lu, Z. Zhang, X. Chen, Y. Lan, Sci. Rep. 4 (2014) 7597. |
| [10] |
Q. Yao, Z.H. Lu, Y. Wang, X. Chen, F. Gang, J.Phy.Chem.C 119 (2015) 14167-14174. DOI:10.1021/acs.jpcc.5b02403 |
| [11] |
Z.H. Lu, J. Li, A. Zhu, et al., Int. J. Hydrogen Energy 38 (2013) 5330-5337. DOI:10.1016/j.ijhydene.2013.02.076 |
| [12] |
D. Nazimek, A. Machocki, T. Borowiecki, Adsorp.Sci.Technol. 16 (1998) 747-757. DOI:10.1177/026361749801600907 |
| [13] |
Šarapatka T.J., Chem. Phys. Lett. 212 (1993) 37-42. DOI:10.1016/0009-2614(93)87104-B |
| [14] |
J.P. Jacobs, L.P. Lindfors, J.G.H. Reintjes, O. Jylhä, H.H. Brongersma, Catal. Lett. 25 (1994) 315-324. DOI:10.1007/BF00816311 |
| [15] |
Q. Yao, Z.H. Lu, H. Wei, X. Chen, Z. Jia, J. Mater. Chem. A 4 (2016) 8579-8583. DOI:10.1039/C6TA02004F |
| [16] |
J. Sehested, J. Catal. 223 (2004) 432-443. DOI:10.1016/j.jcat.2004.01.026 |
| [17] |
J.B. Claridge, M.L.H. Green, S.C. Tsang, et al., Catal. Lett. 22 (1993) 299-305. DOI:10.1007/BF00807237 |
| [18] |
Z. Liu, Y. Wang, J. Li, RSC Adv. 4 (2014) 13280. DOI:10.1039/c3ra46352d |
| [19] |
G. Feng, Ganduglia-Pirovano M.V., C.F. Huo, J. Sauer, J. Phys. Chem. C 122 (2018) 18445-18455. DOI:10.1021/acs.jpcc.8b03764 |
| [20] |
F. Loviat, I. Czekaj, J. Wambach, A. Wokaun, Surf. Sci. 603 (2009) 2210-2217. DOI:10.1016/j.susc.2009.04.032 |
| [21] |
A.N. Fatsikostas, X.E. Verykios, J. Catal. 225 (2004) 439-452. DOI:10.1016/j.jcat.2004.04.034 |
| [22] |
M.A. Keane, G. Jacobs, P.M. Patterson, et al., J. Colloid Interface Sci. 302 (2006) 576-588. DOI:10.1016/j.jcis.2006.06.057 |
| [23] |
X. Li, D. Li, H. Tian, Appl. Catal. B-Environ. 202 (2017) 683-694. DOI:10.1016/j.apcatb.2016.09.071 |
| [24] |
R.P. Galhenage, H. Yan, S.A. Tenney, N. Park, G. Henkelman, et al., J. Phys. Chem. C 117 (2013) 7191-7201. DOI:10.1021/jp401283k |
| [25] |
K.R. Hahn, A.P. Seitsonen, M. Iannuzzi, J. Hutter, ChemCatChem 7 (2015) 625-634. DOI:10.1002/cctc.201402906 |
| [26] |
H. Ay, Üner D., Appl. Catal. B-Environ. 179 (2015) 128-138. DOI:10.1016/j.apcatb.2015.05.013 |
| [27] |
Y. Sekine, D. Mukai, Y. Murai, et al., Appl. Catal. A-Gen. 451 (2013) 160-167. DOI:10.1016/j.apcata.2012.11.005 |
| [28] |
D. Mei, V.A. Glezakou, V. Lebarbier, et al., J. Catal. 316 (2014) 11-23. DOI:10.1016/j.jcat.2014.04.021 |
| [29] |
K.Y. Koo, S.H. Lee, U.H. Jung, H.S. Roh, L.Y. Wang, Fuel Process. Technol. 119 (2014) 151-157. DOI:10.1016/j.fuproc.2013.11.005 |
| [30] |
S.M.D. Lima, A.M.D. Silva, L.O.O.D. Costa, et al., Appl. Catal. A-Gen. 377 (2010) 181-190. DOI:10.1016/j.apcata.2010.01.036 |
| [31] |
G. Li, C. Gu, W. Zhu, et al., J. Clean Prod. 183 (2018) 415-423. DOI:10.1016/j.jclepro.2018.02.088 |
| [32] |
Y. Liu, S. Qing, X. Hou, et al., ChemCatChem 24 (2018) 5698-5706. |
| [33] |
G. Kresse, J. Furthmüller, Comput. Mater. Sci. 6 (1996) 15-50. DOI:10.1016/0927-0256(96)00008-0 |
| [34] |
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [35] |
S. Grimme, S. Ehrlich, L. Goerigk, J. Comput. Chem. 32 (2011) 1456-1465. DOI:10.1002/jcc.v32.7 |
| [36] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [37] |
P.E. Blöchl, C.J. Forst, J. Schimpl, Bull. Mater. Sci. 26 (2003) 33-41. DOI:10.1007/BF02712785 |
| [38] |
P.E. Blöchl, Phys. Rev. B 50 (1994) 17953-17979. DOI:10.1103/PhysRevB.50.17953 |
| [39] |
Q.J. Liu, Z.T. Liu, Appl. Phys. Lett. 99 (2011) 091902. DOI:10.1063/1.3630131 |
| [40] |
O'Neill H.S.C., M. James, W.A. Dollase, S.A.T. Redfern, Eur. J. Miner. 17 (2005) 581-586. DOI:10.1127/0935-1221/2005/0017-0581 |
| [41] |
R.F. Cooley, J.S. Reed, J. Am. Ceram. Soc. 55 (1972) 395-398. DOI:10.1111/jace.1972.55.issue-8 |
| [42] |
X. Yu, X. Zhang, X. Tian, S. Wang, G. Feng, Appl. Surf. Sci. 324 (2015) 53-60. DOI:10.1016/j.apsusc.2014.10.056 |
| [43] |
L. Shi, D. Wang, X. Yu, et al., Mol. Catal. 468 (2019) 29-35. DOI:10.1016/j.mcat.2019.02.009 |