 2019, Vol. 30
2019, Vol. 30 


b School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China;
c Jiangsu Key Laboratory of Drug Discovery for Metabolic Disease, China Pharmaceutical University, Nanjing 210009, China;
d Department of Biochemistry and Molecular Biology, Nanjing Medical University, Nanjing 10029, China
Cancer is one of the major diseases that threaten human health. For decades, people have been committed to the development of highly effective, low-toxic anti-cancer drugs [1]. Many of the anticancer agents were derived from natural products either directly or indirectly [2, 3]. PTX has been widely used in the treatment of breast carcinoma, ovarian carcinoma, part of head-neck cancer and lung cancer [4-6]. In contrast to anti-mitotic drugs, PTX is the first discovered antitumor drug to interact with microtubule polymer and restrain dissociation of tubulin, thereby forcing cell mitosis remained in G2/M period and promoting apoptosis [7, 8]. However, because of the low water solubility, the clinical application of traditional paclitaxel injection requires a mixture of Cremophor EL and absolute ethanol as a solubilizer which could cause a series of allergic reactions and make patients painful [9, 10]. Besides, in clinical application, PTX could cause severe toxicity and adverse reactions as it is a kind of cytotoxic drug lacking targeted specificity, such as neutrophil reduction, allergic reaction, neurotoxicity and hair loss, muscle pain, etc. [11-13]. Therefore, it is necessary to develop new drug delivery systems of paclitaxel which do not contain Cremophor EL and have tumor-targeting specificity to facilitate clinical use and alleviate patient suffering [14-18].
When the concentration reaches critical micelle concentration (CMC) of amphiphilic block copolymer, micelles are formed by selfassembly as a type of drug carrier which could realize controlling release and increase solubility of drugs [19-21]. So, if choose micelles as the drug delivery system for PTX, the problem of poor solubility can be solved and the use of Cremophor EL is avoided. The remaining problem is the targeting specificity of micelles. Because the rapid growth of the tumor cells, the lining of blood vessels of most tumor tissues are damaged making its permeability better than normal tissues. As a result, when polymer material (molecular weight greater than 70 kDa) or nanoparticles (grain diameter between 50~200 nm) go through tumor sites, impaired lymphatic circulation system will have no filtration effect on them, they can be effectively retained near the tumor to release drugs slowly and improve drug targeting performance [22-25]. On the contrary, it is hard for micelles to stay in normal tissues. In addition, microenvironments in the tumor are so different from normal tissues in some aspects that can be beneficial for controlling release of drugs, such as tumor extracellular pH is 6.5–7.2 and intracellular pH is 4.5–6.5 [26, 27]. If the selected micelle carrier is sensitive to the acid environment, when micelles are remained near the cancer and entre the cancer cells, the carrier will degrade and drugs will be released rapidly [28-31].
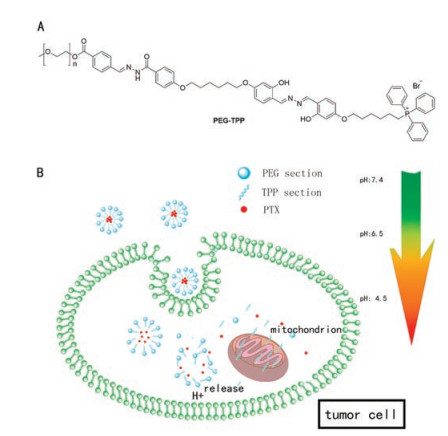
Based on the design concepts above, we synthesized a pHsensitive carrier (PEG-TPP) [32] to envelop PTX in the form of micelles. The structure of PEG-TPP is shown in Scheme 1A, the PEG segment is hydrophilic and the TPP section is hydrophobic, overall PEG-TPP could be dissolve in water, so emulsion solvent evaporation method was used to prepare micelles [33]. As displayed in Scheme 1B, When PTX-loaded micelles approach the tumor site, they are trapped there on account of EPR effect described above and enter cells through endocytosis. Then due to the acidic pH value intracellular, the hydrazone bonds in the structure of PEGTPP will disrupt leading to the release of PTX. Besides, at the end of hydrophobic section of PEG-TPP, triphenyl phosphine (TPP) structure is capable of targeting cell mitochondria [34, 35]. Membrane potential of mitochondria is much higher than other organelles owing to ATP synthesis and ion transport, therefore, TPP as a kind of lipophilic cationic compound could be effectively targeted in mitochondria [36]. Mitochondria play an important role in cell survival and death as they not only produce the ATP providing energy for cell metabolism, but also regulates metabolism and programmed cell death [37]. Mitochondrial dysfunction is a priority for many anti-tumor drugs, and some of them directly interfere with the mitochondrial respiratory chain, eventually leading to cancer cell death. Therefore, theoretically the carrier itself is kind of helpful to improve the anti-tumor activity of PTX.

|
Download:
|
| Scheme 1. (A) the structure of PEG-TPP; (B) PTX-loaded micelles can be taken in tumor cells, and then release PTX, free PEG-TPP targets mitochondria. | |
{kind=link}
In general, we recommend acid-sensitive, fluorescent and antitumor PEG-TPP as a suitable carrier for a new drug delivery system of PTX. The characterizations of PTX-loaded micelles were evaluated, including hydrodynamic diameter, polydispersity, microscopic morphology, CMC, in vitro stability and in vitro release. At the cellular level, we investigated the cellular uptake, the ability to target mitochondrial and in vitro antitumor activity of the PTX-loaded micelles. In vivo experiments, we demonstrated that PTX-loaded micelles have excellent tumor-targeting specificity. The antitumor activity of PTX-loaded micelles was significantly improved compared to paclitaxel alone.
PEG(2000)-OH (700 mg, 0.3 mmol), Br-TPP (265 mg, 0.3 mmol) and K2CO3 (43 mg, 0.3 mmol) were dissolved in 30 mL acetonitrile stirring at 60 ℃ for 24 h and monitored by thin layer chromatography (TLC) performed on GF/UV 254 plates and were visualized using UV light at 254 nm and 365 nm. PEG(2000)-OH and Br-TPP were synthesized as described in our previous study. After reaction completion, acetonitrile was removed in vacuum. The residue was purified by flash column chromatography on silica gel using dichloromethane/methanol (50:1) as eluent to obtain desired product PEG-TPP. Potassium carbonate (K2CO3) and dichloromethane were purchased from Sinopharm Chemical Reagent Co., Ltd. Methanol, acetonitrile and acetone was obtained from Shanghai Titan Scientific Co., Ltd. All solvents were used without further purification unless otherwise indicated.
PTX-loaded micelles are prepared by emulsion solvent evaporation method: 0.5 mg PTX and 10 mg PEG-TPP are co-dissolve in 1 mL acetone, then add it drop by drop into 10 mL water, keep it stir overnight at room temperature. PTX was purchased from Saen chemical technology Co., Ltd. Blank micelles were prepared in the same way without PTX. The mean size (hydrodynamic diameter) of PTX-loaded micelles was measured using particle size instrument (Brookhaven, USA). The particle size measurements were carried out in triplicate. The morphological evaluation was treasured by transmission electron microscopy (TEM, HITACHI, HT7700, Japan) through copper grid following staining with phosphomolybdic acid and infrared grying. Pyrene was used as a fluorescence probe to determine the CMC value of micelles [38]. Typically, pyrene was added into the micelles solutions at concentrations ranging from 10-10 mg/mL to 1 mg/mL and the fluorescent intensity ratio of I394/I373 (I1/I3) was recorded together with the concentrations of micelles to get the CMC value.
After the PTX-loaded micelles are prepared, the stability of PTX-loaded micelles was determined by recording the particle size, encapsulation efficiency and drug loading capacity once a day after one week of incubation. The testing micelles were stored at 4 ℃. After centrifuging, demulsification with methyl alcohol and filtering, samples were detected through HPLC with LC-20 A chromatograph system (SHIMADZU, Japan) to get the content of PTX in micelles. The encapsulation efficiency and loading capacity of PTX were calculated using the following equations:

|
(1) |

|
(2) |
The in vitro release of PTX from micelles was investigated by dialysis method. 1.5 mL micelles were placed in each dialysis bags (MW = 7000, Biosharp, USA) surrounding by 45 mL buffer solution including 1% tween 80 with different pH values: 7.4, 6.5, 4.5. Tween 80 (Aladdin) was used as nonionicsurfactant for PTX to dissolve in buffer solution. After incubating at 37 ℃ and shaking at 100 rpm, 1 mL samples were taken out from the outer buffer solution in each dialysis bags and 1 mL fresh buffer solution was added as compensation at specific times. All the samples were prepared in 48 h and ultimately detected by HPLC. The data analysis and chart generation were performed in GraphPad Prism 5.0.
At the cellular level, the cellular uptake ability of PTX-loaded micelles was observed on K562 cell line and the mitochondria targeting trial was taken on MCF-7 cell line by laser confocal fluorescence microscopy (LCFM, FV1000, Olympus, Japan). K562 cells were maintained in RPMI1640 medium supplemented with 10% fetal calf serum (FBS, Gibco) in a humidified incubator at 37 ℃ with 5% CO2. K562 cells (1 ×105) were seeded on each observation dish and incubated for 12 h at 37 ℃, 5% CO2. Then, add the PTX-loaded micelle at final concentration of 1 mg/mL into the medium of the cells. After incubating at 37 ℃ with 5% CO2 for 1 h, 2 h, 4 h and 8 h, corresponding treatment observation dishes were washed with PBS for three times and then fluorescence imaged by LCFM with the excitation and emission wavelengths being set at 380 nm and 530 nm. MCF-7 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS in a humidified incubator at 37 ℃ with 5% CO2. MCF-7 cells were seeded on observation dishes at the density of 105 cells per dish incubating for 24 h. The cells were then treated with PTX-loaded micelles for 4 h. The cells in dishes were washed with PBS three times and stained by Mito Tracker Red (50 nmol/L) for mitochondria in culture media for 45 min. After staining, the cells were washed with PBS three times and fixed with the treatment of 10% formalin at 4 ℃ for 15 min following imaged by LCFM.
What is more, several tumor cell lines (K562, MCF-7 and Hela) were used to evaluate the in vitro cytotoxicity of PTX, blank micelles prepared by PEG-TPP and PTX-loaded micelles by MTT assay. Hela cells were maintained in DMEM supplemented with 10% FBS in a humidified incubator at 37 ℃ with 5% CO2. Tumor cells were seeded into a 96-well plate at a density of 104 cells per well. After incubation for 24 h, the cells were treated with PTX, blank micelles and PTX-loaded micelles. 48 h later, the medium was removed and MTT dye (10 μL of 5 mg/mL in PBS) was added to each well 4 h prior to experiment. The plates were then centrifuged at 1500 rpm for 15 min and the supernatant was gently discarded. 100 mL DMSO solution was added into each well to dissolve the purple crystals and the plates were softly shaken for 5 min at room temperature. The optical density (OD) was read on a microplate reader (Thermo, USA) with a wavelength of 490 nm.
All animal experimental protocols were approved by the guidelines for the Care and Use of Laboratory Animals of China Pharmaceutical University and conducted according to the Laboratory Animal Management Regulations in China. And they were all adhered to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication NO. 85-23, revised 2011). Athymic nude mice (18–22 g, female) were purchased from Cavens Laboratory Animal (Jiangsu, China). MCF-7 tumor cells were subcutaneous injected into the female athymic nude mice (n = 5) to get the MCF-7 tumor model [39]. As the diameter of the tumor was up to 1 cm, 100 μL of micelle (5 mg/mL) was injected through the vena caudalis. After 4 h, some major organs (heart, liver, spleen, lung, kidney and tumor) of the vehicle and micelle-treating MCF-7 tumor-bearing mice were excised and detected by living imaging system (PerkinElmer, IVIS Spectrum, USA) to observe the distribution of the micelle at the tissue level. What is more, all the excised organs were fixed with 10% buffered formalin and embedded in paraffin. The sliced organs (8 mm) were observed by a BX41 bright field microscopy (Olympus) in order to further confirm the distribution of the micelle at the tissue level.
Then, the U87 tumor model was established to evaluate the therapy activities of PTX, blank micelles and PTX-loaded micelles as a comparison. And the used ICR mice (18–22 g, female) were purchased from Comparative Medicine Centre of Yangzhou University (Jiangsu, China). U87 cells were vaccinated and bred in the ascites of healthy mice (n = 5) following subcutaneously injecting them into the female normal mice (n = 48). When the tumors were suitable in size, the mice were randomly arranged into four groups (n = 12) respectively injected with PBS, PTX, blank micelles and PTX-loaded micelles (200 μL, 5 mg/kg) though the vena caudalis. Administration frequency was once every two days and the treatment period sustained two weeks. In order to quantitatively analyze the effectiveness of different treatments (vehicle, blank micelles generated by PEG-TPP, PTX alone and PTX-loaded micelles), the tumor volume, survival rate, and body weight of each group of mice were recorded at different times during 14- day.
The reaction was showed in Fig. S1 (Supporting information), nuclear magnetic resonance (NMR) spectroscopy were recorded in DMSO-d6 on a Bruker ACF-300Q Instrument (300 MHz for 1H). The results were performed in Fig. S2 (Supporting information), additional hydrogen NMR signals of PEG-TPP belong to Br-TPP. The hydrogen with chemical shift near 11 ppm were phenolic hydroxyl hydrogen of Br-TPP, when chemical shifts ranged from 6 ppm to 8 ppm and from 1 ppm to 3 ppm, the areas represented the aromatic hydrogen and aliphatic hydrogen of Br-TPP respectively.
The characterizations of PTX-loaded micelles were carried out to evaluate their external properties. As shown in Fig. S3 (Supporting information), the polydispersity index was 0.09 ± 0.046 indicating a well-distributed state of micelles and the mean particle size (hydrodynamic diameter) was 152.1 ±1.2 nm. The morphology of fresh-prepared PTX-loaded micelle could be intuitively observed by TEM. The micelles had a regular spherical morphology and the diameter was around 140 nm close to 152.1 nm. The evaluated CMC of PEG-TPP was 7 ng/L by recording the fluorescent intensity ratio of I394/I373 (I1/I3) of Pyrene and corresponding concentrations of PEG-TPP.
After 0, 1, 2, 3, 4, 5, 6, 7 day of micelles incubation, we collected micelles samples to determine the particle size, encapsulation efficiency and drug loading efficiency of PTX-loaded micelles. According to the results in Fig. 1, the hydrodynamic diameter almost remained unchanged, besides, the degrees of decline in encapsulation efficiency and loading capacity did not exceed 50%, so the micelles were proved to be stable in one week since they were prepared. Furthermore, it is important for the micelles which aim at targeting cancer to be sensitive to different pH physiologically. In Fig. 1C, the in vitro release results showed the obvious pH dependence of PTX releasing from micelles, as acidic pH triggering nearly total drug release over a relatively short period. 95% encapsulated drug was released by 48 h at pH 4.5 and nearly 60% was released in first 4 h. In contrast, when pH was 6.5 or 7.4, nearly 50% or 30% was released by 48 h in a gentle way respectively.

|
Download:
|
| Fig. 1. (A) The particle size of PTX-loaded micelles during the 7 days; (B) The encapsulation efficiency as histogram and loading efficiency in line graph of PTX in micelles; (C) PTX release from copolymer micelles at pH 4.5, 6.5 and 7.4. | |
{kind=link}
In vitro cellular uptake of PTX-loaded micelles was evaluated to determine whether the micelles can enter the cancer cells and how fast they can get inside (Fig. S4 in Supporting information). After 1 h incubation of K562 cells with micelles, almost no green fluorescence was observed in the cells. After 2 h, faint fluorescence was observed. After 4 h, strong fluorescence was observed. After 8 h, the fluorescence intensity was almost the same as that at 4 h.
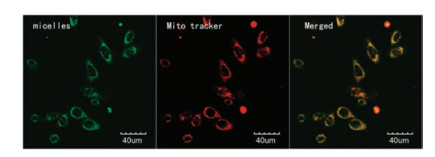
MCF-7 cell line was used to investigate the targeting cell mitochondria capacity of micelles bearing a TPP structure (Fig. 2), after 4 h treated with micelles and then Mito-Tracker Red. Micelles and Mito-Tracker Red emit green and red fluorescence, respectively. The merged image of red fluorescence and green fluorescence appearing as the yellow fluorescence in the cytoplasm of the treated cells indicates the successful mitochondria-targeted delivery of micelles.

|
Download:
|
| Fig. 2. Green fluorescence of micelles and red fluorescence of mito tracter appered at same place in cells. | |
{kind=link}
At the cellular level, the anti-tumor activity of blank micelles, paclitaxel, and PTX-loaded micelles was measured by MTT assay on three tumor cell lines. The in vitro cytotoxicity testing was carried out in triplicate. The cytotoxicity results of blank micelles, paclitaxel, and PTX-loaded micelles are shown in Table S1 (Supporting information). The results on the three cell lines all indicated that the cell killing power of the PTX-loaded micelles was stronger than that of the blank micelles and paclitaxel, especially in the MCF-7 cell line, IC50 value of PTX-loaded micelles was 123.2 nmol/L less than a half of that of PTX (298.3 nmol/L). PEGTPP itself exhibited weak cytotoxicity effect on all tested cell lines.
PEG-TPP can emit green fluorescence, so after injection of micelles in vivo, it is possible to speculate on the tissue distribution of the micelles by observing fluorescence. Some major organs (heart, liver, spleen, lung, kidney and tumor) of the vehicle and micelle-treating MCF-7 tumor-bearing mice were excised and detected by living imaging system, green fluorescence was only detected in tumor (Fig. 3B). Furthermore, the sliced organs (8 mm) were observed by a BX41 bright field microscopy (Olympus) in order to further confirm the distribution of the micelle at the tissue level (Fig. 3C).

|
Download:
|
| Fig. 3. (A) PTX-loaded micelles emits green fluorescence at the excitation and emission wavelengths being set at 380 nm and 530 nm; (B) Excised organs in micelles treating group are detected at same condition; (C) Microscopic images of freezing microtome section of excised organs in micelles treating group. | |
{kind=link}
PTX-loaded micelle has a circular appearance and an appropriate diameter which is considered to play a pivotal role in tumor targeting based on EPR effect. The CMC value of PTX-loaded micelles is 7 ng/L, generally speaking, when the CMC is lower, the more predicted particles will be formed which can be more advantageous for the hydrophobic drugs to be loaded in the hydrophobic inner side at the same polymer concentration. PTX-loaded micelles were proved to be stable in one week since they were prepared and possess strong pH dependence. So, theoretically, after injection of the micelles, they will keep stable and integral in normal tissues, when they are remained in the tumor and contact with the acidic microenvironments, the micelles will rupture and release a large amount of paclitaxel to act on tumors.
At the cellular level, the micelles can enter the K562 cells achieving a balance in 4 h. When micelles enter the MVF-7 cells, part of them or debris including TPP fragment are capable of locating mitochondria and interfere with the normal function of the mitochondria, causing cell apoptosis, which may be benefit to the anti-tumor effect of paclitaxel because mitochondria not only produce ATP to provide energy for cell survival, but also participate in programmed cell death. This is proved by cytotoxicity of PTX and PTX-loaded micelles on the three cell lines, IC50 values of PTX-loaded micelles were much lower than that of PTX alone.
Restricted by poor penetrability of green fluorescence, the distribution of micelles in living treated mice failed to be observed in vivo imaging system. The fluorescent distributions of both isolated organs show that the micelles are capable of targeting solid tumor, In accordance with integral organs, intense green fluorescence was observed in sliced tumor, what is more, weak fluorescence occurred in sliced kidney indicating part of micelles have metabolized through kidney, while other organs in micelletreating mice and vehicle group appeared to be non-blooming. All in all, it was obvious that PTX-loaded micelles can target solid cancer effectively.
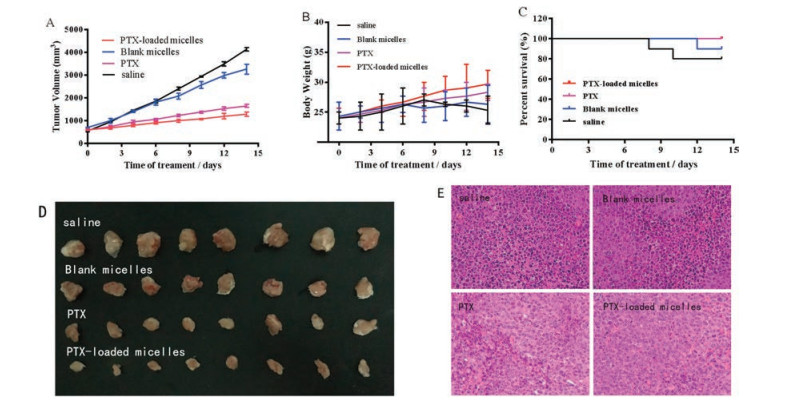
Based on the successful validation of the in vivo targeting efficacy and in vitro antitumor activity, we next investigated whether PTX-loaded micelle could enhance therapeutic efficacy in mice bearing U87MG tumors. As illustrated in Fig. 4D, no obvious therapeutic effects were observed in blank micelles-treated mice, implying that PEG-TPP was equipped with sort of anticancer ability. Mice treated with PTX-loaded micelles exhibited obvious benefits (versus mice treated with saline), which was even better than PTX. Tumor volume belonging to the mice treated with saline and blank micelles are much larger than the mice treated with PTX or PTX-loaded micelles (Fig. 4A). Body weight and survive rate usually reflect the health condition of the treated mice. As shown in Fig. 4B, the body weight of mice in the control group and blank For the PTX-loaded micelles group treated group, their body weight gradually increased during 14 days, implying that systemic toxicity was minimal in these mice. The mice treated with PTX and PTX-loaded micelles maintained a 100% survival rate after 14 days, which is higher than the mice treated with blank micelles (90%) and saline (80%) as shown in Fig. 4C. To further compare the anticancer efficiency in different treating groups, ex vivo histology studies were performed (Fig. 4E). Significant necrosis occurred in the peripheral and central tumor region for PTX group, and only a small amount of tumor cells appeared around the tumor vessels. For PTX-loaded micelles group, it is more obvious that there was hardly good looking tumor cell could be observed in the section where cell morphology was not clear and the cancer cells have been badly damaged indicating a better antitumor activity of PTX-loaded micelles than PTX alone. As for blank micelles and vehicle groups, widespread well-shaped and melanocratic cancer cells could be observed, especial the vehicle group, which means therapeutic effect of blank micelles was weak. What is more, there was no apparent difference made out in the sections of other organs (heart, liver, spleen, lung, kidney) from different treating groups (Fig. S5 in Supporting information).

|
Download:
|
| Fig. 4. (A) Tumor growth curves of the U87MG-tumor-bearing mice after intravenous injection with saline, blank micelles, PTX and PTX-loaded micelles. *P < 0.05; (B) Body weight of mice in different treatments groups during 14 days; (C) Survival rate of mice in different treatment groups. *P < 0.05; (D) Images of the tumors in different treatment groups obtained after the 14-day therapy; (E) Images of tumor after H&E staining examined by histological analysis. | |
{kind=link}
On the whole, we developed pH-responsive and fluorescent micelles on the basis of PEG-TPP which can effectively deliver and release Paclitaxel into cancer cells so as to enhance the anticancer activity. The pH-responsive and fluorescent micelles are suffice stable and capable of controlling drug release under physiological conditions that about 30% PTX was released by 48 h at pH 7.4, 37 ℃.The results show that PTX-loaded micelles have the ability to locate mitochondria inside the cancer cells and interfere with mitochondrial function in consideration of the modest cytotoxicity of blank micelles. Besides, both in vitro and in vivo, the anticancer ability of micelles were superior to PTX alone. For in vivo improved anticancer ability, the contribution of targeting cancer capacity cannot be neglected as fluorescent micelles distribute mainly in cancer rather than other organs. These pHresponsive and fluorescent micelles appeared to be degradable, selective and potentially strengthened platform for traditional antineoplastic drugs which may be transformed into potent nanomedicines.
AcknowledgmentsThis study was supported by grants from the National Natural Science Foundation of China (No. 81872733) and the Natural Science Foundation of Jiangsu Province of China (No. 15KJB310004).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.01.003.
| [1] |
L.A. Torre, F. Bray, R.L. Siegel, et al., CA-Cancer J. Clin. 65 (2015) 87-108. DOI:10.3322/caac.21262 |
| [2] |
K.H. Altmann, J. Gertsch, Nat. Prod. Rep. 24 (2007) 327-357. DOI:10.1039/B515619J |
| [3] |
G.M. Cragg, P.G. Grothaus, D.J. Newman, Chem. Rev. 109 (2009) 3012-3043. DOI:10.1021/cr900019j |
| [4] |
C. Brignone, M. Gutierrez, F. Mefti, et al., J. Transl. Med. 8 (2010) 71. DOI:10.1186/1479-5876-8-71 |
| [5] |
S. Nagao, N. Iwasa, A. Kurosaki, et al., Int. J. Gynecol. Cancer 22 (2012) 70-75. DOI:10.1097/IGC.0b013e318234f927 |
| [6] |
R. Vashistha, J.S. Sethi, H. Singh, et al., Int. J. Cancer (2002) 427. |
| [7] |
X. Ling, R.J. Bernacki, M.G. Brattain, F. Li, J. Biol. Chem. 279 (2004) 15196-15203. DOI:10.1074/jbc.M310947200 |
| [8] |
A.F. Wahl, K.L. Donaldson, C. Fairchild, et al., Nat. Med. 2 (1996) 72-79. DOI:10.1038/nm0196-72 |
| [9] |
J.Y. Yamaguchi, Y. Nishimura, A. Kanada, et al., Toxicology 211 (2005) 179-186. DOI:10.1016/j.tox.2004.10.019 |
| [10] |
C. Khanna, M. Rosenberg, D.M. Vail, J. Vet. Intern. Med. 29 (2015) 1006-1012. DOI:10.1111/jvim.2015.29.issue-4 |
| [11] |
G.A. Orr, P. Verdier-Pinard, H. McDaid, S.B. Horwitz, Oncogene 22 (2003) 7280-7295. DOI:10.1038/sj.onc.1206934 |
| [12] |
F.E. Walker, Semin. Oncol. Nurs. 9 (1993) 6-10. DOI:10.1016/S0749-2081(16)30036-5 |
| [13] |
F. Salvinelli, M. Casale, B. Vincenzi, et al., J. Exp. Clin. Cancer Res. 22 (2003) 155-158. |
| [14] |
Y.L. Luo, M. Han, F. Xu, Y.S. Chen, Y.Q. Zhang, Macromol. Biosci. 15 (2015) 1411-1422. DOI:10.1002/mabi.v15.10 |
| [15] |
D.J. Zhao, H.Y. Zhang, S.F. Yang, W.X. He, Y.X. Luan, Int. J. Pharmaceut. 515 (2016) 281-292. DOI:10.1016/j.ijpharm.2016.10.029 |
| [16] |
T. Zhong, X. Yao, S. Zhang, et al., Sci. Rep-UK. 6 (2016) 36614. DOI:10.1038/srep36614 |
| [17] |
X. Chen, X. Ling, L. Zhao, et al., ACS. Appl. Mater. Inter. 10 (2018) 33976-33985. DOI:10.1021/acsami.8b11571 |
| [18] |
J. Wang, W. Xu, S. Li, et al., J. Biomed. Nanotechnol. 14 (2018) 2102-2113. DOI:10.1166/jbn.2018.2624 |
| [19] |
H. Cho, T.C. Lai, K. Tomoda, G.S. Kwon, AAPS PharmSciTech 16 (2015) 10-20. DOI:10.1208/s12249-014-0251-3 |
| [20] |
A. Gothwal, I. Khan, U. Gupta, Pharm. Res. 33 (2016) 18-39. DOI:10.1007/s11095-015-1784-1 |
| [21] |
P.P. Constantinides, M.V. Chaubal, R. Shorr, Adv. Drug. Deliver. Rev. 60 (2008) 757-767. DOI:10.1016/j.addr.2007.10.013 |
| [22] |
V. Torchilin, Adv. Drug. Deliver. Rev. 63 (2011) 131-135. DOI:10.1016/j.addr.2010.03.011 |
| [23] |
C. Yang, S.Q. Liu, S. Venkataraman, et al., J. Control. Release 208 (2015) 93-105. DOI:10.1016/j.jconrel.2015.03.027 |
| [24] |
W. Li, J. Peng, L. Tan, et al., Biomaterials 106 (2016) 119-133. DOI:10.1016/j.biomaterials.2016.08.016 |
| [25] |
X. Chen, L. Zhao, Y. Kang, et al., Front. Pharmacol. 9 (2018) 118. DOI:10.3389/fphar.2018.00118 |
| [26] |
R. Cheng, F.H. Meng, C. Deng, Z.Y. Zhong, Nano Today 10 (2015) 656-670. DOI:10.1016/j.nantod.2015.09.005 |
| [27] |
W. Chen, P. Zhong, F. Meng, et al., J. Control. Release 169 (2013) 171-179. DOI:10.1016/j.jconrel.2013.01.001 |
| [28] |
Z. Wang, X. Deng, J. Ding, et al., Int. J. Pharmaceu. 535 (2018) 253-260. DOI:10.1016/j.ijpharm.2017.11.003 |
| [29] |
P. Hassanzadeh, F. Atyabi, R. Dinarvand, J. Control. Release 270 (2018) 260-267. DOI:10.1016/j.jconrel.2017.12.007 |
| [30] |
Y. Hao, W. Li, X. Zhou, F. Yang, Z. Qian, J. Biomed. Nanotechnol. 13 (2017) 1581-1597. DOI:10.1166/jbn.2017.2474 |
| [31] |
Y. Qu, B. Chu, K. Shi, J. Peng, Z. Qian, J. Biomed. Nanotechnol. 13 (2017) 1598-1618. DOI:10.1166/jbn.2017.2475 |
| [32] |
J. Li, Y. Liu, H. Li, et al., Biomater. Sci. 6 (2018) 2998-3008. DOI:10.1039/C8BM00889B |
| [33] |
J. Yu, H. Deng, F. Xie, et al., Biomaterials 35 (2014) 3132-3144. DOI:10.1016/j.biomaterials.2013.12.074 |
| [34] |
J. Hou, X. Yu, Y. Shen, et al., Nanoscale Res. Lett. 12 (2017) 158. DOI:10.1186/s11671-017-1931-1 |
| [35] |
C.M. Paleos, D. Tsiourvas, Z. Sideratou, Mol. Pharmaceut. 13 (2016) 2233-2241. DOI:10.1021/acs.molpharmaceut.6b00237 |
| [36] |
Q. Hu, M. Gao, G. Feng, B. Liu, Angew. Chem. 53 (2014) 14225-14229. DOI:10.1002/anie.v53.51 |
| [37] |
C.C. Hsu, L.M. Tseng, H.C. Lee, Exp. Biol. Med. 241 (2016) 1281-1295. DOI:10.1177/1535370216641787 |
| [38] |
D.G. Ahn, J. Lee, S.Y. Park, Y.J. Kwark, K.Y. Lee, Appl. Mater. Inter. 6 (2014) 22069-22077. DOI:10.1021/am505444c |
| [39] |
H. Chen, Y. Di, D. Chen, et al., Nanoscale 7 (2015) 8884-8897. DOI:10.1039/C5NR00473J |