 2019, Vol. 30
2019, Vol. 30 


b Department of Chemical and Biomolecular Engineering, The University of Akron, Akron OH 44325, United States;
c State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China;
d School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China;
e Nanjing Daniel New Mstar Technology Ltd., Nanjing 211200, China;
f National Demonstration Center for Experimental Biomedical Engineering Education, Southeast University, Nanjing 210096, China
Breast cancer is the most common invasive cancer in women, and the second main cause of cancer death in women. The symptoms and the need for screening are important ways of reducing the risk. Quick and simple screening method for breast cancer needs a rapid development. Microfluidic chip and SELEX (systematic evolution of ligands by exponential enrichment) technology were present new strategies for rapid diagnosis and treatment for breast cancer cells.
In recent decades, the microfluidic chip has been widely used in life science, disease diagnoses and treatment, environmental monitoring, drug synthesis and screening and has been one of the most popular frontier technologies [1-3]. The microfluidic chip is known as chip laboratory when the reactions were done on the chip [4]. Cell chip is an important type of chip laboratory according to integrate different cells onto a single chip for different biochemical operations in order to get a precise diagnosis of potential diseases. The developments of cell chip provide a new insight into drug screening [5]. Cell chips have advantages in cell culture as providing a suitable microenvironment, ability to realize the cell culture, cell operation and cell analyze. In last decade, useing of microfluidic cell chip technology for trace cell capture, culture, and biological research has become an important application [5, 6]. Some work combined the microfluidic cell chip with immunomagnetic to separate different cell lines [7]. But for breast cancer, gastric cancer, and colon cancer cell lines that cannot be screened by traditional immunomagnetic separation because they do not express epithelial secretion factor Ep-CAM (Epithelial Cellular Adhesion Molecule) [8]. How to make the cell chips for breast cancer keep in stable performance? Providing a reliable micro-environment and effectively target of cell-lines will be very important for the development of cells-growing chips.
SELEX (systematic evolution of ligands by exponential enrichment) is a high-throughput biological library screening technology and brought new ideas for wide range tumor marker [9-12]. By SELEX technique the aptamer as an "artificial antibody", have many advantages over protein-based antibodies, such as being immune to the restriction of immunogenicity, accurate in recognition, small molecules, and easy penetration and so on [13-15]. Tan's team keep in studying with SELEX screened to enabling the specific identification of target tumor cells from clinical specimens. Aptamer obtained by an abatement of SELEX technology can be further used to capture its target substance, thereby obtaining specific tumor biomarkers [16]. However, a strategy to effectively apply SELEX technique or use DNA segment to screen tumor cells is still a challenge in this field [17].
In our research, a cell microarray chip system based on DNA segment modification and 3D embedded cell scaffold were designed to enables accurate, rapid diagnosis and target treatment of breast cancer cells SK-BR-3 in vitro. In the system, cancer cells were cultured in a hydrogel-based 3D environment to form a cell microarray. We developed a micro fluidic device to perform a continuous cell culturing by replacing culture medium automatically. By controlling the diffusion of small molecules, cell microarray chip system can also screen drugs targeting into SK-BR-3 cells. Based on the thiol aptamers target with breast cancer cells, we designed a visualized cell microarray for breast cancer cell screening. Experimental results show that the device can achieve drug screening and detection functions, and has very broad commercial application prospects.
We explore different reaction concentration to choose the most appropriate reaction system. We dissolve sodium alginate using ultrapure water as a solvent, making 0.01~0.03 g/mL sodium alginate (SA) ultrapure water solutions. Using DMSO or mixture of ultrapure water and DMSO to make 0.01~0.03 g/mL calcium chloride (CaCl2) solutions. The ratio of ultrapure water and DMSO is 1:1~2:1. The sodium alginate solution reacts with calcium chloride solution in a flat-bottomed container with the ratio around 1:1 in a vacuum drier with the temperature below 16- degree Fahrenheit and the environment pressure below -0.1 MPa for more than 10 min. We prepared 3 react systems (Table 1): 1% SA solution, 2% CaCl2 (ultrapure water:DMSO = 1:1) solution; 1% SA solution, 1% CaCl2 (DMSO) solution; 1% SA solution, 3% CaCl2 (DMSO) solution.
|
|
Table 1 Sample composition and material appearance. |

Six well cell culture plates were used as the cell culture rooms. The microfluidic injection device was used to control the flow of the drug.
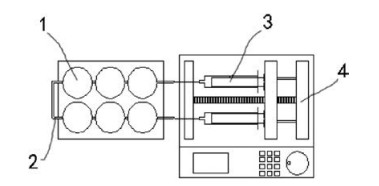
The chip is consisted of cell culture plate, the details as show in Fig. 1: (1) circular cell culture rooms; (2) 0.5 mm diameter tube with a micro valve (black diamond); (3) syringe; (4) injection speed is controlled by microinjection pump. Two units connect with cell rooms separately and the drug is injected at a rate of 2 μL/s into the reaction vessel; three-dimensional ultrafine porous cell scaffold is placed in each cell culture room.

|
Download:
|
| Fig. 1. Model of cell microarray chip system. | |
DNA segment for SK-BR-3 can specifically combine with the target molecule in cell according to bases pairing principle [18]. Strategy of designing DNA segment for SK-BR-3 is based on overexpressing of the HER2/c-erb-2 gene product [19]. By Marking DNA segment with fluorescein isothiocyanate (DAF), we can detect signal through fluorescence microscope and realize the detection of the target molecule on SK-BR-3. DNA segments marked with fluorescein isothiocyanate (FITC) and covalent bonding with Camptothecin (DAF-CPT) [20] target to SK-BR-3 were designed and applied to different cell lines to evaluate the chip's screening effect on SK-BR-3 cells.
The sequence of the DNA aptamer for SK-BR-3 is based on overexpressing of the HER2/c-erb-2 gene product and shows as following: CACTACAGAGGTTGCGTCTGTCCCACGTTGTCATGGGGGG TTGGCCTGTTTTTT.
Scanning electron microscope was used to observe our 3D cell scaffold materials to observe the materials' surface, porosity, and uniformity distribution of pores to ensure that the physical properties of the material meet the basic needs of cell experiments.
Mechanics testing equipment Instron 2752–005 tensile testing apparatus (Instron, MA, USA) was used to stretch the strips with a steady slow force until strips break. Hydrogel (width-lengththickness: 15 mm–30 mm–1 mm) were used as gel samples. Film strips of sodium alginate hydrogel (30 × 15 mm) were held between two pneumatic clamps separated at a distance of 15 mm and pulled apart with a constant cross head speed of 20 mm/min. Tensile strength and elongation at break were calculated using BlueHills software (Instron, MA, USA). The specimen was stretched until the gel fractured and the fracture extension of the gel specimen was recorded as Lc. The fracture energy were expressed as Eq. (1):
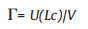
|
(1) |
where Γ is the fracture energy, U(Lc) is work done by the force at the displacement of 0-Lc calculated from the area below the forcedisplacement curve, V were the volume of gel specimen.
Preweighed, sodium alginate hydrogel samples (1 cm2 square pieces) were placed into Eppendorf tubes filled with 2 mL sterile PBS (0.1 mol/L, pH 7.4) containing penicillin (100 units/mL) and streptomycin (100 μg/mL) then incubated (37 ℃, 150 rpm). At periodic intervals hydrogel samples were removed, filtered (2 μm pore size) with deionised water to capture all the particulate pieces and freeze dried (-60 ℃, 48 h) then left to stand in a clean laminar flow hood for moisture equilibration (25 ℃, RH 25%, 12 h). The samples were subsequently weighed and weight loss (WL) expressed as percentages of the initial. As shown in Eq. (2):
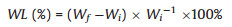
|
(2) |
where Wf and Wi are the final and initial weights respectively. Means of 5 replicates were determined (n = 5).
We used Annexin V-FITC (sigma) to test the cell apoptosis in cell model after drug delivery. Cell scaffold was cut into 1 cm2 circular pieces and sterilization. 6 pieces of cell scaffold were lay down into 6 well plates separately to form as a cell chip. Cultivate the treatment and control groups of human breast cancer cell line (SKBR-3), human liver cancer cell line (HepG2) and human aortic smooth muscle cells (HASMC) separately in 6 rooms of the cell chip. Each type of cell has two rooms marked as #1 and #2, #2 rooms were given 20 μg/mL CPT, #1 rooms were with culture medium. After 24 h culture, the cell chips were washed twice with PBS, when introduce to analysis, sterile PBS were used for both #1 and #2 rooms. For cell apoptosis assay, 105 cells (three types separately) were gathered and suspended in binding buffer. The cell aliquots were then stained with 5 μL Annexin-V-FITC and 5 μL Propidium Iodide and incubated for 10 min in darkness. Then, the apoptotic induction was assessed by flow cytometer.
Three types of cells HepG2, HASMC, SK-BR-3 were cultured in six different cell culture rooms on the chip. After 24-h culture in incubator with continuously cell culture medium supplementary, three rooms in one channel were supplied with cell culture medium and DNA-aptamer with drugs, as treatment group the other three rooms in the other channel were supplied with only cell culture medium as control group. We observed them under laser confocal microscope.
For confocal microscopy observation, cells were washed twice with PBS. Cells were observed under confocal laser scanning microscope (TCS SP8, Leica, Germany), λex 488 nm were applied.
Cell scaffold was cut into 1 cm2 circular pieces and soaked in 75% alcohol. Sterilize chip and scaffold under UV light for 30 min. Put 6 pieces cell scaffold into chip and use sterilized PBS wash for 3 times and soak in PBS for 1 h. Get cell lines: SK-BR-3, HepG2, HASMC cultivate them in 6 cell culture rooms. Each type of cell has two rooms marked as #1 and #2. #1 rooms were given 500 μL free fluorescent dye and reacted 30 min and #2 rooms were given 20 μg/mL DAF-CPT. After 24-h culture, cells were washed and soaked with PBS. fluorescence microscope observe were applied. Trypan blue were used for calculate death cells.
Cell microarray chip is very similar to 6 well cell culture plate, cells were cultured on the scaffold in the culture rooms of the chip. The microfluidic injection device was used to control the flow of the drug. As shown in Fig. 2, microinjection pump can control the velocity of flow range from 0.01 mL/h to 1 mL/h. We placed 1 cm2 striped 3-dimensional porous cell scaffold (inside picture of Fig. 2) in each 6-cell culture room and cultured 3 types of cell. As shown in blue color channel in Fig. 2, DAF-CPT was designed as one integrate to use as fluorescence tracer and targeting drug treatment for SKBR-3. Blue color is from trypan blue used for calculating death cells. DNA segment marked by fluorescence (DAF) was used for control group without drug treatment as shown in another channel (transparent) in Fig. 2.

|
Download:
|
| Fig. 2. Cell microarray chip system. | |
DAF-CPT is DNA segments marked with FITC and covalent bonding with camptothecin. It is a type of DNA aptamer in our experiment. This type of DNA aptamer can specifically and selectively combine with SK-BR-3 targeting matter, used as a probe in detection and assay [21]. DAF-CPT in our cell micro-array chip showed a good sensibility as antigen-antibody reaction and meantime better stability. With suppling of DAF-CPT and controllable culture-medium continuously, the cell micro-array chip will be very useful for observing and targeting treatment for SK-BR-3. As shown in Fig. 2, by using trypan blue analysis, we can evaluate the surviving of cells in an effective way.
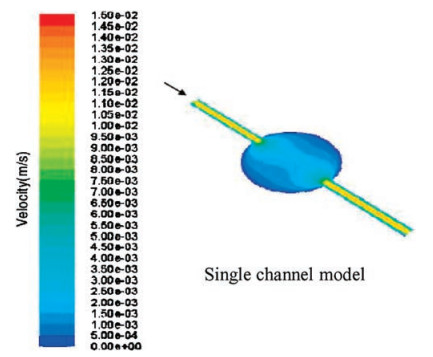
Our device is featured as a fast disposable device, so the chip materials need to be cheap and accessible. The diameter of tough tubes is 0.5 mm. As shown in Fig. 3, when the cell culture rooms are circular and the tubes are connected, flowing culture solution and drugs have the shear force below the damage threshold at the same time. The changing rate of liquids in the room is fast enough to avoid liquid residue.

|
Download:
|
| Fig. 3. Distribution of velocity. | |
It has been widely reported that sodium alginate can have crosslinking reaction with calcium chloride and produce sodium alginate gel and used for cells adhesion [22]. This gel's performances such as hardness differ with different reaction concentration, amount of calcium combination, environment conditions when react. In this work, we used a mixture of different ratios of SA, CaCl2, and DMSO as raw materials for cell scaffolds. The above three kinds of materials have all passed the test for cytotoxicity in vitro and have proved to have good biocompatibility and are level 0. Meanwhile, SA is very common in biological laboratory for example cell culture hydrogels. We use SA hydrogel as our 3D cell scaffold and DMSO for pore former on the gel.
Because of the DMSO's high volatility, it is very easily turn into gas during the cross-linking reaction. The material was placed into the vacuum drying oven keep in temperature of 60 ℃. In vacuum, DMSO volatilized sharply and formed pores in the gel. In the meantime, because of DMSO's high volatility, it is not likely leave residual in the gel and reduced its possible damage to cell.
As shown in Table 1 and Fig. 4A (sample 1), when the ratio of component was 1% SA, 2% CaCl2 (pure water):DMSO = 1:1, pores on the surface of the material can be found penetration and uniform. When the ratio of component was 1% SA mixed with 1% CaCl2 in DMSO, the pores on the surface of the material (sample 2) are tiny and show a sparse quantity. When the ratio of component was 1% SA mixed with 3% CaCl2 that dissolved in DMSO, fold on the surface of sample 3 can be found. As a 3D cell scaffolds, the materials should allow recapitulation of the extracellular environment of cells by providing attachment sites, the ability for cells to grow in 3D shape. Based on this phenomenon, it can be speculated that sample 1 can be used as cell scaffolds

|
Download:
|
| Fig. 4. (A) SEM imaging of sample 1, sample 2 and sample 3. (B) Degradation percentage curve of sample 1, sample 2, sample 3. (C) Fracture energy evaluation of sample 1. | |
We also used alcohol instead of DMSO in sample 1 based on its good volatility. Observe by optical microscope and SEM no pores or fold can be found on the surface of the materials. This type of materials cannot use as 3D cell scaffold. It is because the DMSO owns higher vapor pressure than alcohol.
The SEM imaging results are shown in Fig. 4A. It can be seen from the SEM image of sample 1 that pores on the surface of the material can be found penetration and uniform dispersed on the surface of the material. However, the surface of the sample 2 has less pores. Different from samples 1 and 2, the uneven surface of material can be found on sample 3.
The SEM images also confirmed that this sodium alginate gel of sample 1 has pore and the porosity is greater than 90%, the pore size is around 0.1~1 μm. The material with even ultra-small pore may facilitate the cells' mutual communication. Also, this kind of porous materials may promote cell culture as a cell scaffold and facilitate the cells' adhesion. Unlike traditional two-dimensional cell scaffold, a three-dimensional cell scaffold may facilitate cell grows into three dimensions and imitating 3D environment in vivo helps imitate real tissue in human body.
Cell three dimensional is a crucial step in imitating real tissue in vivo. This cell scaffold may also facilitate the cells' mutual communication and material exchange such as many growth factors and pheromone. It may promote drug's permeation and increase the contact rate between cells and drug.
Mechanics characteristics of hydrogels will be important for cell scaffold, one with good elasticity and not easily to be broken will be ideally using for cell culture. The other important property of the materials is degradation velocity. Our chip is designed for fast screening, usually tested within 24 h, the biodegradation velocity of materials should meet the requirement. In next step, mechanics characteristics and biodegradation velocity of materials has been analysed.
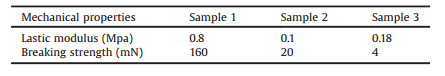
Cell scaffold material requires enough breaking strength and need to be flexible for cell cultivating. Stretching test applied on the cell scaffold using a biomechanics measurement platform to test the elasticity modulus and breaking strength of the materials. As can be found from Table 2, the modulus of elasticity of sample 1 was 0.8 MPa and the breaking strength was 160 mN. In comparison with all other two samples, sample 1 was good elasticity and not easily to be broken.
|
|
Table 2 Elastic modulus and breaking strength. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fracture energy is another indicator of the toughness of the hydrogels. Fig. 4C is a typical force-extension curves of at mm/mm (tension rate) for a tensile fracture test. As we can see in this figure when displacement was at 14 mm, the peak force was 160mN, it meant that the hydrogel could achieve a fracture energy at 0.35 J/m2. The results show that if the displacement of the arms is less than 14 mm, the load of sample 1 increases with the increase of displacement and can reach the top velum of 160mN. From the fracture energyofhydrogel as show in the results, we deduced thatin the reaction system of sample1, SA-CaCl2 ionic bond crosslinkers formed more completely when they were mixture in DI water. The fracture energy of the hydrogels can reach to 0.35 J/m2 (Eq. (1)). The fracture energy shows sample 1 was good elasticity and not easily to be broken. This type of materials will be good for cell growth.
In conclusion, the elastic modulus and the fracture energy of sample 1 is the largest during all of the three different types of materials. It is well known that with small elastic modulus, the material will show week elastic deformation. At the same time greater the fracture energy is, greater the tensile strength of the material is. We deduced from the experiments that penetration holes on sample 1 will result a change in shape of a material at low stress that is recoverable after the stress is removed. The complete reaction of SA and CaCl2 supplied strong ionic interaction force and allow the materials have high breaking strength and fracture energy, these characters of materials will provide the most suitable environment for organ cells growth, such as SK-BR-3 and HepG2.
The first order kinetic degradation reaction is the reaction rate inversely proportional to the primary side of the reactant content in the system. Mathematical model for the differential equation:
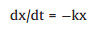
|
(3) |
where t is the time; x = x (t) is the content of the reactants in the system; the first derivative dx/dt is the reaction rate; the ratio coefficient k is the reaction rate constant, k > 0, the negative sign indicates the reactant content in decay. Mathematical models of first-order kinetics have many applications, such as radioactive decay, heating or cooling, absorption and exclusion of drugs in the human body, and degradation of pollutants.
We also conducted Materials degradation test on samples 1, 2, 3. As shown in the Fig. 4B, before 72 h, the percent degradation curve of sample 1 satisfying physical properties is consistent with the first order kinetic degradation curve; and after 72 h, the degradation curve collapses. From the results we found that before 72 h, curve of degradation shows gentle and progressive, the phenomenon indicated that a small number of glycosidic bonds of SA and ionic bonds of SA-CaCl2 were breaking but the whole hydrogel networks were still stable. The breakage of the glycosidic bonds of alginate (AG) and ionic bond of SA-CaCl2 reached a critical value after 72 h, decreasing of the curve shows that the whole crosslinking network disjointed, fragmented network present in the whole hydrogel.
Based on the experimental results of physical characterization and elasticity measurement, we choose sample 1 as a cell scaffold for later biological experiments. Cell cycle experiments was less than 48 h, the collapse of sample 1 after 72 h will not affect the growth of different types of cells. From the apoptosis results as show in Figs. 5A and 6A (Without drug), all of the three types of cells do not show prominent process of programmed cell death when they were cultured on sample 1. From apoptosis analysis, the early and late apoptosis cells are all in a low level and over 98% cells show a good vitality in all three types of cells. From the confocal image, all three types of cells are in a good morphology. We conformed from the results that scaffold sample 1 is in low toxic and compatible with all the three types of different cells. Combining with the penetration and uniform pores on the surface (Fig. 4A) and mechanical property (Fig. 4C), this scaffold will supply a three dimensions (3D) tissue-like environment for cells and will be suitable used as biological devices of cell micro-array chip. Compared with traditional cell culture, this cell scaffold is more beneficial to cell growth and buffer transport or cells communication. Thus, this model may imitate real tissue environment in vivo. Because of precise peristaltic pump, the model can supply liquid continuously and quantitatively. The throughout channels can be procyclical or countercyclical as required. The device can imitate in vivo environment conveniently by inside 3D porous cell scaffold and continuous cell culture medium supplementary. The transparent upper cover makes it easy to conduct multiple biological assays such as fluorescence analysis band detection. Based on the above advantages of our cell micro-array chips, this cell test model may realize the drug highly efficient screening and multi-cells typing.

|
Download:
|
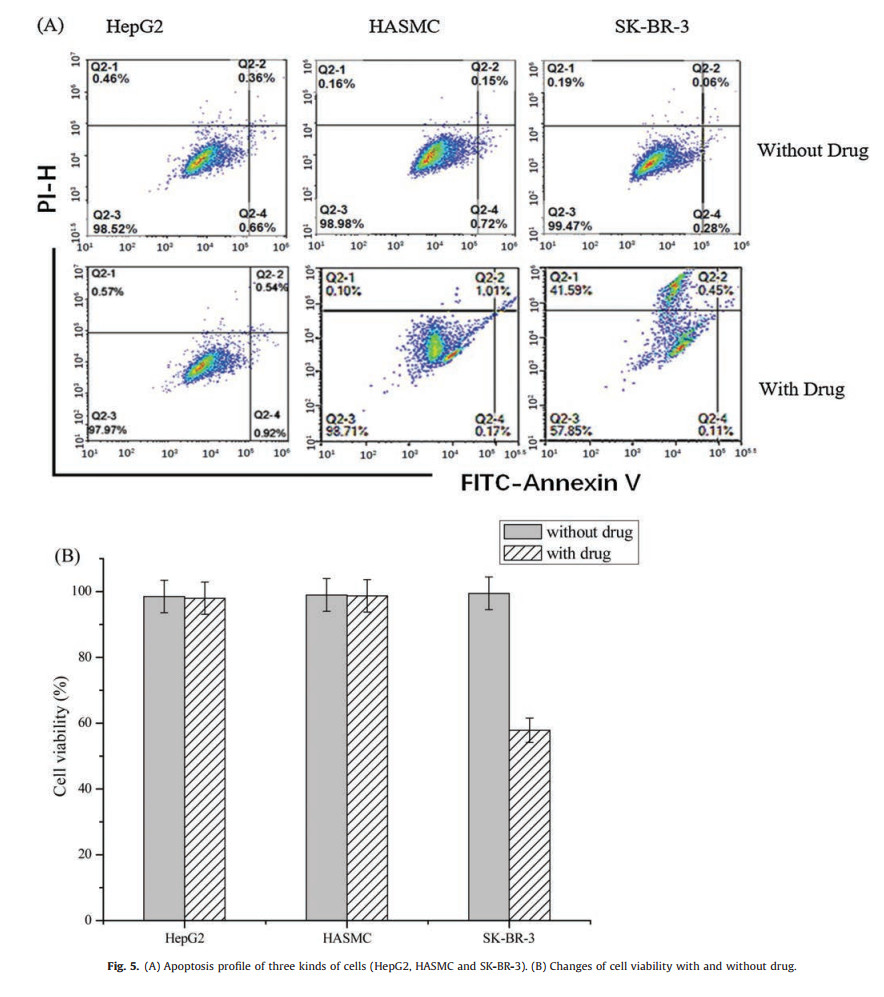
| Fig. 5. (A) Apoptosis profile of three kinds of cells (HepG2, HASMC and SK-BR-3). (B) Changes of cell viability with and without drug. | |
{kind=link}

|
Download:
|
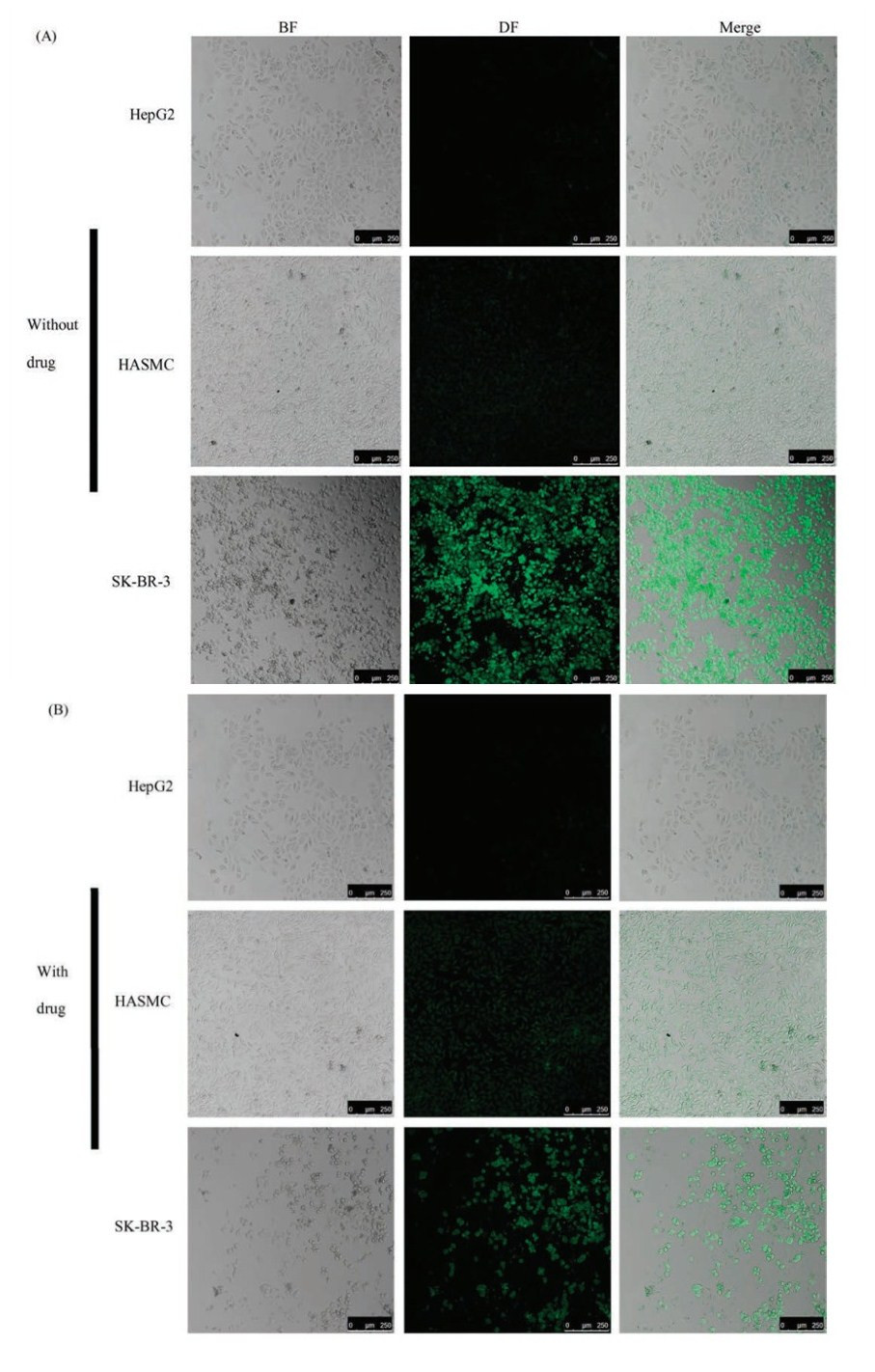
| Fig. 6. Confocal images of the cells in the chip marked with DAF or DAF-CPT (A) Cells were cultured with DAF (without drug) (B) Cells were cultured with DAF-CPT (with drug). | |
{kind=link}
After fabrication of whole microfluidic device, we conducted cell apoptosis assay to evaluate the apoptosis in chip. Cell apoptosis is one of the main indicators evaluating the drug's targeting and eliminating effect. Fluorescent dyes may bind or intercalate with different cellular components such as DNA or RNA to evaluate the cancer drug's performance by comparing the treatment and control groups. We applied DAF and DAF-CPT to the three different types of cells in the cell micro-array chip to evaluate the chip's screening effect on cancer drug.
As shown in Fig. 5A (without drug), when DAF flow through one side of the cells micro-array chip, the cells early apoptosis or late apoptosis cannot be found in all of the three different types of cells. After the DAF-CPT treatment in the other micro-array chips (Fig. 5 with drug), large quantities of early apoptosis can be found in SKBR-3 cell lines, while, for the HepG2 and HASMC cells, small quantities of late apoptosis can be found in the same channel. By analyzing bar graph in Fig. 5B, we found that when targeting by DAF-CPT, the survivor of human liver cells HepG2 were found been slightly influenced by camptothecin during 24 h. Similar as HASMC cells, 1% decreasing of viability cells can be found with treatment of DAF-CPT. Differently, quantities of living SK-BR-3 cells sharply decreased, only 58% survival cells can be found after the treatment of DAF-CPT. The sharp decreasing of bar graph clearly shows the targeted killing of SK-BR-3 cells by DAF-CPT, while, unchanging of bar graph for other two types of cells show that HepG2 and HASMC do not influence by DAF-CPT.
Apoptosis test results demonstrates that with the micro-array cell chip, DAF-CPT can effectively target and kill SK-BR-3 cells but does not work for HASMC and HepG2 cells. Cell micro-array chip with model cells that cultured in cell rooms which contain 3D porous cell scaffold can be used for evaluate the therapeutic efficacy in a very rapid and simple way. By supplying drug and cell culture medium continuously and quantitatively, our cell microarray chip can realize the drug screening.
To further confirm cell micro-array chip can realize the drug screening, the different channels treated with DAF or DAF-CPT were observed by confocal laser scanning microscope. DAF or DAFCPT was design to specifically combine with the biotin in breast cancer cell, ideally only the breast cancer cell can be marked by FITC.
As show in Fig. 6A, all the three types of cells were first observed by bright field microscope to describe the cells morphology then in dark field for observation of fluorescence from DAF. From the bright field (BF) and merged image, it is clearly show that without drug treatment, human cells HepG2, HASMC and SK-BR-3 cell line are all in large density after 24 h of cells culture. Dark field (DF) images shown that bright green fluorescence can be found in cell room of SK-BR-3 cell line but week fluorescence signals can be found in HepG2 and HASMC cell lines. Results indicates that DAF cannot combine with HepG2 and HASMC cells.
After treated with DAF-CPT for 24 h as show in Fig. 6B, the human breast cancer cells SK-BR-3 can be found in bright green fluorescence but small quantities. At the same time, SK-BR-3 cells can be found crack and split after treated with the targeting drugs, just little quantity of cells still in healthy situation. However, HepG2 and HASMC cell lines were still in a great density of cells, at the same time cell morphology of the two cell lines displayed that after treatment of DAF-CPT, cells were still in a healthy situation.
SK-BR-3 cells in Figs. 6A and B distinct differences in cell density and shape, which indicates that the drug has strong influence and damage on SK-BR-3 cells. The results demonstrate that in our cell micro-array chip, drug can work effectively on the cells cultivated in cell room with inside 3D porous cell scaffold and the drugs were delivered by precise peristaltic pump and work on cells through throughout channel between cell culture rooms. Meanwhile, by our cell micro-array chip, drug screening can be realized by confocal microscope to distinguish the cell density and morphology change with drugs. Specifically, in this experiment, only the SKBR-3 cells were marked by DAF or DAF-CPT and show fluorescence signal. Results indicate that the cell microarray chip designed in our work has great targeting and lethality to SK-BR-3 cells. And our cell micro-array chips can be intuitive and quickly distinguish SKBR-3 cells. Therefore, our work was conducive to detection of SKBR-3 cell in unhealthy organ, which can realize specific detection of SK-BR-3 cells.
In this work, we successfully constructed a cell micro-array chip based on microfluidic device with an inside three-dimensional porous cell scaffold. This device can cultivate normal cells and cancer cells in three-dimensional environment and supply continuously with cell culture medium and drug, which imitating real environment in vivo is benefit for cells' communication and material transport. We used a fast and simple method to manufacture a gel based on sodium alginate with ultra-small pore can be used as an inside 3D cell scaffold as its good mechanical property. The mechanical strength of 3D cell scaffold is similar to organ in vivo. So, the hydrogel of SA-CaCl2 canprovide an environment forhealthy growth of cells. We applied this cell scaffold to a microfluidic chip and assemble with microfluidic device to realize continuous liquid supplementary. By using the DNA segments targeting to SK-BR-3 as model drug, we designed DAF and DAF-CPT. By controlling the transport of drugs and medium in cell micro-array chip, SK-BR-3 cells canbe targetedkilled amongdifferent celllines.In addition, the described method enables the production of multiple copies of a chip by using a simple, fast and inexpensive array fabrication without any specialized equipment. The device has a promising future in drug screening and cell detection.
AcknowledgmentsThis research is supported by the National Natural Science Foundation of China (Nos. 11204033, 51773093), the Natural Science Foundation of Jiangsu Province (No. BK20141397), the Research Fund for the Doctoral Program of Higher Education of China (No. 20120092120042), the CMA L'OREAL CHINA SKIN GRANT 2015 (No. S2015121421) and the Open Research Fund of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. SKLNMKF201803), Southeast University and Nanjing Medical University Cooperation Project (No. 2242018K3DN14), Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University.
| [1] |
R. Hersher, Nat. Med. 18 (2012) 475-475. DOI:10.1038/nm0412-475 |
| [2] |
K.G. Lee, T.J. Lee, S.W. Jeong, et al., Sensors 12 (2012) 10810-10819. DOI:10.3390/s120810810 |
| [3] |
M. Niimi, T. Masuda, K. Kaihatsu, et al., Sens. Actuators B-Chem. 201 (2014) 185-190. DOI:10.1016/j.snb.2014.04.011 |
| [4] |
S. Park, H.S. Moon, D.S. Lee, et al., Int. J. Lab. Hematol. 3 (2013) 480-490. |
| [5] |
K. Pu, C. Li, N. Zhang, et al., Biosens. Bioelectron. 89 (2016) 927-931. |
| [6] |
T. Huang, C.P. Jia, W.J. Sun, et al., Biosens. Bioelectron. 51 (2014) 213-218. DOI:10.1016/j.bios.2013.07.044 |
| [7] |
M. Tang, C.Y. Wen, L.L. Wu, et al., Lab Chip 16 (2016) 1214-1223. DOI:10.1039/C5LC01555C |
| [8] |
T. Meyaard, A. Vries, T. Ruiter, et al., J. Exp. Med. 198 (2003) 1129-1135. DOI:10.1084/jem.200309031941107cor |
| [9] |
M. Ye, J. Hu, M.Y. Peng, et al., Int. J. Mol. Sci. 13 (2012) 3341-3353. DOI:10.3390/ijms13033341 |
| [10] |
O. Shoji, Biores. Open Access 1 (2012) 265-272. DOI:10.1089/biores.2012.0253 |
| [11] |
Y.M. Kim, C. Liu, W.H. Tan, Biomark. Med. 3 (2009) 193-202. DOI:10.2217/bmm.09.5 |
| [12] |
M. Chen, Y. Yu, F. Jiang, et al., Int. J. Mol. Sci 17 (2016) 2079. DOI:10.3390/ijms17122079 |
| [13] |
M. Svobodová, A. Pinto, P. Nadal, et al., Anal. Bioanal. Chem. 404 (2012) 835-842. DOI:10.1007/s00216-012-6183-4 |
| [14] |
K.T. Guo, P. Angela, S. Christian, et al., Int. J. Mol. Sci. 9 (2008) 668-678. DOI:10.3390/ijms9040668 |
| [15] |
H. Sun, Y. Zu, Molecules 20 (2015) 11959-11980. DOI:10.3390/molecules200711959 |
| [16] |
K.F. Blount, R.R. Breaker, Nat. Biotechnol. 4 (2006) 1558-1564. |
| [17] |
Z.X. Huang, Q. Xie, Q.P. Guo, et al., Chin. Chem. Lett. 28 (2017) 1252-1257. DOI:10.1016/j.cclet.2017.01.002 |
| [18] |
T. Kato, R. Shimada, M. Kimura, et al., Chem. Commun. Lett. 52 (2016) 4041-4044. DOI:10.1039/C5CC08816J |
| [19] |
K. Dastjerdi, G.H. Tabar, H. Dehghani, A. Haghparast, Biotechnol. Appl. Biochem. 58 (2011) 226-230. DOI:10.1002/bab.36 |
| [20] |
H. Fujita, M. Kuwahara, Curr. Protoc. Nucleic Acid Chem 65 (2016) 9.10.1-9.10.19. |
| [21] |
H.L. Cai, C.S. Zhou, Q. Yang, et al., Chin. Chem. Lett. 29 (2018) 531-534. DOI:10.1016/j.cclet.2017.09.010 |
| [22] |
D.W. Zhu, Z. Chen, et al., Chin. Chem. Lett. 26 (2015) 807-810. DOI:10.1016/j.cclet.2015.04.033 |