 2019, Vol. 30
2019, Vol. 30 


b Key Laboratory of Metabolomics at Shenzhen, Shenzhen 518055, China;
c Department of Hepatobiliary and Pancreatic Surgery, Shenzhen People's Hospital, Second Clinical Medical College of Jinan University, Shenzhen 518000, China;
d Shenzhen Kivita Innovative Drug Discovery Institute, Shenzhen 518110, China
Metastasis is the major cause of cancer-related deaths in the world, which involves a multistep cascade of biological events, specifically stromal invasion and vascular invasion. Tumorassociated macrophages (TAMs), the most abundant population of immune cells in tumor microenvironment, are key promoters for tumor metastasis and therapeutic resistance [1-3]. Clinical studies have shown that monocytes can be recruited by some malignant tumor cells, and infiltrating macrophages in tumor tissue are associated with tumor grades and poor prognosis [4, 5]. These TAMs have the ability to enhance tumor cell invasion and migration by secreting of chemokines, cytokines, growth factors, and metabolites [6, 7]. Moreover, interstitial flow (IF), driven by pressure differences between blood, interstitial and lymphatic compartments, is an important biophysical cue in tumor microenvironment, and it plays a prominent role in tumor progression [8, 9]. Many researches have demonstrated that elevated tumor IF in the tumor stroma leads to mechanical strain and spatial gradients of secreted cytokines to promote angiogenesis and tumor invasion.
Recently, microfluidic technology has become a powerful tool for cell-based researches [10-12]. Its obvious advantages involve flexible cell manipulation, quantitative and controlled modulation of cell-cell microenvironment, and compatible with many analytical techniques, which allow real-time and dynamic cellbased study [13-16]. And the technology to generate 3D breast tumor microenvironment on a chip is maturing. Until now, there are several researches about separate effect of macrophages and IF on tumor cell migration and invasion on microfluidics. They found that macrophages had the ability to induce endothelial barrier impairment and tumor intravasation, and promote the dispersion of lung carcinoma aggregates via intercellular cell adhesion molecule-1 (ICAM-1) and β2 integrin interaction [17, 18]. Moreover, macrophages had been reported to release tumor necrosis factor α (TNFα) and transforming growth factor β1 (TGFβ1) to enhance both speed and persistence of cancer cell migration [19, 20]. Although these works were innovative, they ignored the influence of tumor cells on the macrophage recruitment and activation by the secreted cytokines and chemical compounds, which played important roles in tumor progression. Moreover, the crosstalk between macrophages and endothelial cells in tumor microenvironment was still rarely studied on these platforms. Although the effect of IF on tumor cell migration, invasion and angiogenesis was investigated on microfluidics in a controlled way [21, 22], the combined effects of IF and macrophages on tumor cell behaviors remain largely unknown. The comprehensively understanding will help us better understand tumor progression and provide some insights into cancer treatment.
In this work, we developed an in vitro 3D breast tumor model on the microfluidic device to study the interactions between macrophages, tumor cells, and endothelial cells in the presence of IF. The microfluidic device with four-parallel microchannels co-cultured breast cancer aggregates, macrophages, monocytes and endothelial cells within 3D extracellular matrix (ECM). In addition, IF was generated by hydrostatic pressure gradient without additional equipment. Our results indicated that the activated macrophages and IF simultaneously promoted endothelial sprouting and breast cancer aggregates invasion. This assay was the first attempt to reveal the crosstalk between macrophage and tumor aggregates in the presence of IF. The established microfluidic platform can be applied for the mechanism study of macrophages-tumor communication and drug screening.
We firstly constructed a 3D breast tumor microenvironment on a chip. A photo of the fabricated microfluidic device is shown in Fig. 1a. Cancer cells are surrounded by a complex tumor microenvironment consisting of ECM, tumor-associated stromal cells, and vascular endothelial cells. To study the effects of macrophages and IF on the process of tumor cell invasion into surrounding stroma, we constructed a more biomimetic 3D in vitro breast cancer model on a microfluidic device. As shown in Fig. 1b, four parallel-connected channels were designed to co-culture of tumor cells, macrophages and endothelial cells. The widths of the microchannels from left to right were 1.0 mm, 1.2 mm, 1.5 mm, and 1.0 mm, respectively, and the depth of each microchannel was 120 μm (Figs. 1c and d). Two outermost channels were used for cell culture medium supplement and drug stimulation. Collagen gel was employed to suspend human umbilical vein endothelial cells (HUVECs), and the solution was introduced into the microvessel channel to create a 3D microvessel network. After 2 h of gel polymerization, the breast tumor aggregates and U937 monocytes embedded with collagen gel were injected into the tumor channel. In order to generate IF across the central microchannels, hydrostatic pressure gradient with defined volume difference between two reservoirs of outermost fluidic microchannels was applied. IF was verified by measuring flow velocity using 70 kDa FITC-dextran according to previously reported method [23]. In particular, IF generation was simple and easy handling without any specific equipment. As shown in Fig. 1e, IF velocity increased gradually from 0.5 μm/s to 6.5 μm/s when the volume difference varied from 20 μL to 90 μL. The interstitial flow rate generated from the volume difference of 80 μL is similar to that of 90 μL, with the IF velocity of more than 5 μm/s. IF velocities ranged from 0.1 μm/s to 10 μm/s are typically assumed by the use of human cancer models [24]. Therefore, both conditions are suitable for the conditions in cancer microenvironment. We finally selected a volume difference of 80 μL to simulate interstitial flow because of its obvious phenomenon and longer injection time than that of 90 μL.

|
Download:
|
| Fig. 1. Reconstruction of 3D breast tumor microenvironment in the presence of IF on a microfluidic device. (a) An image of the microfluidic device. (b) Schematic of breast tumor microenvironment. (c) and (d) Schematic of the device to mimic natural 3D breast tumor microenvironment, containing breast tumor aggregates, U937 cells, and HUVECs in separate channels with the existence of IF. (e) Relationship between hydrostatic pressure gradient and IF rate. | |
{kind=link}
To investigate the abilities of different phenotypes of breast cancer cells to differentiate U937 monocytes into TAMs, U937 cells were co-cultured with normal breast cells MCF10 A, two typical epithelial-like cancer cells (MCF7 and T47D) and two mesenchymal-like breast cancer cells (MDA-MB-231 and BT549) separately in the adjacent microchannels for 2 days. As shown in Fig. S1a (Supporting information), only when U937 cells co-cultured with MDA-MB-231 or BT549 cells, cell shapes became elongated. A specific macrophage biomarker CD68 was further measured, and the expression images in different co-culture systems were shown in Fig. S1b (Supporting information). These results demonstrated that the mesenchymal-like cancer cell lines had the ability to induce macrophage activation. As reported by numerous studies, CD68 as a total macrophage marker and CD163/CD204 as a M2 macrophage marker were usually detected together to further identify TAMs [25, 26]. Based on this situation, we collected the activated U937 cells from the microfluidic device for RT-PCR assay. As shown in Figs. S1c and S1d (Supporting information), only MDAMB-231 cells could activate the expression of CD68, CD163, and CD204 in U937 monocytes to become M2 macrophages. Taken together, MDA-MB-231 cells could directly activate U937 monocytes towards TAM-like phenotype macrophages.
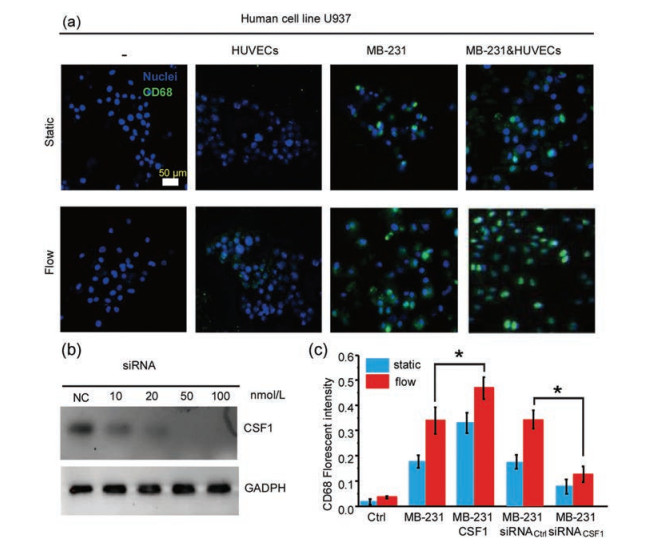
To explore the effect of IF generated in tumor microenvironment on monocyte and macrophage differentiation, U937 monocytes were co-cultured with HUVECs and MDA-MB-231 cells in the adjacent microchannels, respectively. As shown in Fig. 2a, under static condition, only when U937 cells co-cultured with MDA-MB- 231 cells or tri-cultured with MDA-MB-231 cells and HUVECs cells, some of the U937 cells could be differentiated into macrophages, which indicated by the expression of surface biomarker CD68. With the presence of IF, the degree of differentiation was significantly enhanced. The results indicated that MDA-MB-231 cells in the presence of IF could induce differentiation of monocytes into macrophage to a higher extent, compared with that static condition.

|
Download:
|
| Fig. 2. Macrophages activation on the microfluidic device. (a) On-chip fluorescent CD68/DAPI staining of U937 when co-cultured with HUVECs or MDA-MB-231 separately or tri-culture with both cells under static and IF conditions. Scale bar: 50 μm. (b) RT-PCR analysis of colony-stimulating factor (CSF1) excretion from MDA-MB-231 cancer cells transfected with different concentration of specific siRNAs. (c) Influence of CSF1 on the activation of macrophages under different culture conditions, which were evaluated by CD68 staining. | |
{kind=link}
To investigate whether MDA-MB-231 cells activated U937 cells towards TAMs-like phenotype macrophages through CSF1-CSFR signalling pathway, CSF1 gene in MDA-MB-231 cells was knocked down to different extent using specific siRNA. As expected, there was more than 80% knockdown in the gene level for CSF1 with 50 nmol/L siRNA reagent after transfection compared with that nonsilencing control siRNA (Fig. 2b). As shown in Fig. 2c, under static condition, the fluorescence intensity of CD68 in U937 and MDAMB-231 cells co-culture system with the presence of CSF1 was obviously higher than the co-culture system without the presence of CSF1 or with CSF1 gene knockdown. Compared with the same co-culture system in the static condition, the expression levels of CD68 increased to different degrees under the IF condition. The above results revealed that CSF1 played an important role in the activation of monocytes to TAM-like phenotypes by MDA-MB-231 cells.
Due to the important regulators of TAMs and IF in tumor microenvironment, we investigated their combined effects on the angiogenic sprouting using co-culture (HUVECs co-cultured with U937 or MDA-MB-231 cells) and tri-culture (HUVECs tri-cultured with U937 and MDA-MB-231 cells) systems on the microfluidic device. DIO-labelled HUVECs were introduced into the microvessel channel. Other cells were introduced into the tumor channel separately for 3 days culture. As shown in Fig. 3a, IF had the ability to induce the HUVECs sprouting in the opposite direction of IF, which was consistent with previous studies [23, 27]. We also found that MDA-MB-231 cells had the potential to induce endothelial cells sprouting, whereas U937 cells had no contribution on the angiogenic sprouting. When comparing the co-culture with triculture systems, we observed that the sprouting ability of HUVECs into 3D collagen matrix was significantly enhanced in tri-culture system, especially under the IF condition. Endothelial capillary network was further stained by F-actin/DAPI to confirm the above hypothesis. As shown in Fig. 3b, HUVECs sprouted into the adjacent ECM channel in the presence of IF and macrophages were activated obviously.

|
Download:
|
| Fig. 3. Effects of macrophages and IF on tumor angiogenesis. (a) The fluorescence images of HUVECs sprouting under different culture systems. HUVECs stained with DIO were co-cultured with U937 or MDA-MB-231 separately or tri-cultured with both cells on the microfluidic platform for 3 days, respectively. Scale bar: 200 μm. (b) The immunofluorescence images of HUVECs sprouting in 3D collagen matrix in the presence of IF and the macrophages (MΦ). Scale bars: 200 mm. (c) The endothelial microvessel area in the central channel after 3 days cultured with indicated experimental conditions (Bars represent mean ± S.E.M. from at least 3 devices per condition. *** P < 0.001, NS: no sense). αTGFβ1 and αVEGFα stand for anti-TGFβ1 and anti-VEGFα neutralizing antibody. | |
{kind=link}
We further investigated the effects of VEGFα and TGFβ1 on HUVECs sprouting on our established platform. As shown in Fig. 3c, the treatment of cells with VEGFα markedly increased the sprouting vessel area, but treated with TGFβ1 resulted in no significant promotion on the HUVECs sprouting. When the activated macrophages treated with anti-VEGFα neutralizing antibody, HUVECs sprouting almost disappeared, whereas antiTGFβ1 neutralizing antibody had slight influence on the HEVECs sprouting. The above results suggested that VEGFα might be the primary factor involved in the macrophages and IF induced HUVECs sprouting.
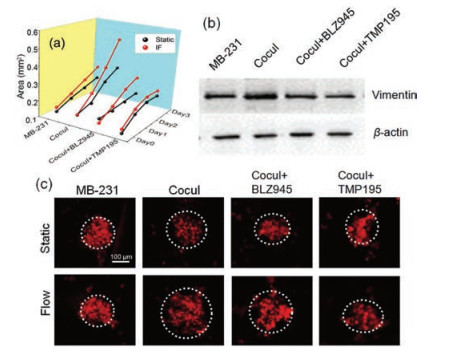
To determine the effects of TAMs and IF on MDA-MB-231 aggregates dispersion, MDA-MB-231 tumor aggregates were cocultured with U937 cells in 3D collagen gel with or without the presence of IF. As shown in Fig. 4a and Fig. S2 (Supporting information), MDA-MB-231 aggregates dispersion were enhanced in the presence of macrophages after 3 days culture. BLZ945, a highly selective CSF1R inhibitor, and TMP195, a novel HDAC Class IIa inhibitor [28], had the abilities to inhibit macrophages activation and reprogram macrophages, thereby affecting tumor growth. When we added the two inhibitors into the co-culture system separately, the results showed that the MDA-MB-231 aggregates dispersion were both significantly suppressed, which were accordance with previous reported results [29]. Vimentin protein, a potential marker related to tumor invasion [30], was further assessed to evaluate the invasiveness of MDA-MB-231 cells by western blotting. As shown in Fig. 4b, MDA-MB-231 cells cocultured with U937 cells expressing higher level of vimentin, the expression of vimentin in MDA-MB-231 cells was down-regulated with the addition of BLZ945 or TMP195 in the co-culture system, which verified that their abilities to inhibit breast tumor cell invasion through inhibiting the activity of TAMs. The degree of the dispersion was characterized by comparing the MDA-MB-231 cell aggregate size (mean of major and minor axis length) to the initial size. Moreover, as shown in Fig. 4c, the dispersion of MDA-MB-231 cells decreased after treated with BLZ945 or TMP195, but the dispersion slightly increased with the present of IF when comparing with static condition. Taken together, the macrophages and IF jointly promoted breast cancer cells dispersion and invasion.

|
Download:
|
| Fig. 4. Effects of macrophages and IF on breast tumor aggregates dispersion and invasion. (a) Quantitative measurement of aggregate dispersion from 0 to 3 days under different culture conditions. (b) Western blotting analysis of vimentin level under different culture conditions with or without drug treatment. β-actin was used as loading control. (c) Corresponding fluorescence images of MDA-MB-231 cell dissemination in the 3D collagen gel. Cocul: co-culture. | |
{kind=link}
In summary, we present a microfluidic-based approach to investigate the effect of macrophages on tumor cell invasion into the surrounding stroma in the presence of IF. The microfluidic device with four-parallel connected microchannels was designed, and could be flexibly used to build a more biomimetic breast cancer microenvironment, containing tumor aggregates, macrophages, monocytes and endothelial cells cultured in 3D collagen matrix. From the results, we proposed a possible mechanism governing the crosstalk of macrophagestumor cells, macrophages-endothelial cells in the presence of IF (Fig. S3 in Supporting information). Firstly, MDA-MB-231 aggregates had the ability to activate monocytes into TAMphenotype like macrophages. IF enhanced the secretion of CSF1 from MDA-MB-231 cells, which further promoted macrophages activation. Secondly, the activated macrophages and IF simultaneously promoted HUVECs sprouting mainly via VEGFα secretion, which enhanced the dispersion and invasion of breast cancer cells. Here, we provide new insights into the crosstalk between macrophages and tumor cells in 3D tumor microenvironment, which has great potential for molecular mechanism study and drug screening assay.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (No. 21675096).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.02.017.
| [1] |
T. Kitamura, B.Z. Qian, J.W. Pollard, Nat. Rev. Immunol. 15 (2015) 73-86. DOI:10.1038/nri3789 |
| [2] |
L. Yang, Y. Zhang, J. Hematol. Oncol 10 (2017) 58. DOI:10.1186/s13045-017-0430-2 |
| [3] |
A. Mantovani, F. Marchesi, A. Malesci, L. Laghi, P. Allavena, Nat. Rev. Clin. Oncol. 14 (2017) 399-416. DOI:10.1038/nrclinonc.2016.217 |
| [4] |
R. Braster, M. Bogels, R.H. Beelen, M. van Egmond, Immunobiology 222 (2017) 21-30. DOI:10.1016/j.imbio.2015.08.011 |
| [5] |
M.D. Sorensen, R.H. Dahlrot, H.B. Boldt, S. Hansen, B.W. Kristensen, Neuropathol. Appl. Neurobiol. 44 (2018) 185-206. DOI:10.1111/nan.2018.44.issue-2 |
| [6] |
K. Sawa-Wejksza, M. Kandefer-Szerszen, Arch. Immunol. Ther. Exp. (Warsz) 66 (2018) 97-111. DOI:10.1007/s00005-017-0480-8 |
| [7] |
N. Linde, M. Casanova-Acebes, M.S. Sosa, et al., Nat. Commun 9 (2018) 21. DOI:10.1038/s41467-017-02481-5 |
| [8] |
M.J. Mitchell, R.K. Jain, R. Langer, Nat. Rev. Cancer 17 (2017) 659-675. DOI:10.1038/nrc.2017.83 |
| [9] |
M.R. Horsman, P. Vaupel, Front. Oncol 6 (2016) 66.. |
| [10] |
M. Rothbauer, H. Zirath, P. Ertl, Lab Chip 18 (2018) 249-270. DOI:10.1039/C7LC00815E |
| [11] |
J. Wu, Q. Chen, W. Liu, Z. He, J.M. Lin, TrAC-Trend. Anal. Chem. 87 (2017) 19-31. DOI:10.1016/j.trac.2016.11.009 |
| [12] |
T. Yuan, D. Gao, S. Li, Y. Jiang, Chin. Chem. Lett. 30 (2019) 331-336. DOI:10.1016/j.cclet.2018.07.013 |
| [13] |
S. Mao, W. Zhang, Q. Huang, et al., Angew. Chem. Int. Ed. 57 (2018) 236-240. DOI:10.1002/anie.201710273 |
| [14] |
C. Lin, L. Lin, S. Mao, et al., Anal. Chem. 90 (2018) 10326-10333. DOI:10.1021/acs.analchem.8b02133 |
| [15] |
M. Jie, H. Lin, Z. He, et al., Sci. China Chem. 61 (2018) 236-242. DOI:10.1007/s11426-017-9167-0 |
| [16] |
M. Jie, S. Mao, H. Li, J.M. Lin, Chin. Chem. Lett. 28 (2017) 1625-1630. DOI:10.1016/j.cclet.2017.05.024 |
| [17] |
C.P. Huang, J. Lu, H. Seon, et al., Lab Chip 9 (2009) 1740-1748. DOI:10.1039/b818401a |
| [18] |
J. Bai, G. Adriani, T.M. Dang, et al., Oncotarget 6 (2015) 25295-25307. |
| [19] |
R. Li, J.D. Hebert, et al., Cancer Res. 77 (2017) 279-290. DOI:10.1158/0008-5472.CAN-16-0442 |
| [20] |
X. Cui, R.T. Morales, W. Qian, et al., Biomaterials 161 (2018) 164-178. DOI:10.1016/j.biomaterials.2018.01.053 |
| [21] |
S. Kim, M. Chung, J. Ahn, et al., Lab Chip 16 (2016) 4189-4199. DOI:10.1039/C6LC00910G |
| [22] |
E. Um, J.M. Oh, S. Granick, Y.K. Cho, Lab Chip 17 (2017) 4171-4185. DOI:10.1039/C7LC00555E |
| [23] |
S. Kim, M. Chung, N.L. Jeon, Biomaterials 78 (2016) 115-128. DOI:10.1016/j.biomaterials.2015.11.019 |
| [24] |
J.M. Munson, A.C. Shieh, Cancer Manage. Res. 6 (2014) 317-328. |
| [25] |
Y. Komohara, Y. Fujiwara, K. Ohnishi, M. Takeya, Adv. Drug Deliv. Rev. 99 (2016) 180-185. DOI:10.1016/j.addr.2015.11.009 |
| [26] |
Y. Komohara, M. Jinushi, M. Takeya, Cancer Sci. 105 (2014) 1-8. DOI:10.1111/cas.12314 |
| [27] |
J.W. Song, L.L. Munn, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 15342-15347. DOI:10.1073/pnas.1105316108 |
| [28] |
L. Cassetta, J.W. Pollard, Cell Res. 27 (2017) 963-964. DOI:10.1038/cr.2017.63 |
| [29] |
J.L. Guerriero, A. Sotayo, H.E. Ponichtera, et al., Nature 543 (2017) 428-432. DOI:10.1038/nature21409 |
| [30] |
A. Satelli, S. Li, Cell. Mol. Life Sci. 68 (2011) 3033-3046. DOI:10.1007/s00018-011-0735-1 |