 2019, Vol. 30
2019, Vol. 30 


Catalytic conversion of alcohols to corresponding carbonyl compounds is an extensively explored research topic in organic synthesis [1-4]. Numerous heterogeneous catalysts have been developed in the past decades [1, 5-12], including various Au, Pd and Pt nanoparticles [10, 13-15]. Among them, nano-scaled Au particles were found to be highly efficient catalysts, as illustrated in the selective oxidation of alcohols into corresponding aldehydes in water [16-18]. Also found was that, monolayer-protected goldpalladium clusters in the polyimide membrane emerge as highly active heterogeneous metal catalysts for solvent-free oxidation of allylic, aliphatic and benzylic alcohols, producing aldehydes and ketones with high selectivity [19]. Alternative designs have also been conceived for advanced, more efficient catalytic systems which do not use molecular oxygen or air as oxidants [20-23]. The development of such promising O2-free methodology is particularly interesting from both practical and environmental points of view since it eliminates the formation of H2O. Note that, in such reactions, a byproduct often deactivates catalysts and necessitates tedious purification of products from the aqueous reaction mixture. Furthermore, it is also tolerant toward alcohols having O2-sensitive functional groups, producing H2 which is an attractive feedstock for energy generation and suppresses over oxidation of the substrate to carboxylic acids.
While small sizes and quantum effect of gold clusters bring forth novel catalysis, the structure stability and reaction dynamics could alter their superior catalytic performance. Recently, utilizing small gold clusters as catalysts, the reactivity and selectivity of glycerol in the conversion to different chemicals have been studied, where the preferentially formed AunO* was found to play a dominant role in the catalytic process for the Jones oxidation [17], as well as glycerol to epichlorohydrin [24]. Insights into the gold cluster catalysis stimulates us a further study here without using oxygen or hydrogen peroxide as primary oxidants. Using a customized setup for laser ablation in liquid (LAL), along with high resolution mass spectrometry and reaction kinetics simulation calculations based on density functional theory (DFT), we have investigated the oxidant-free conversion of glycerol to carbonyl compounds over the small gold clusters. The choice of glycerol is motivated by the fact that it appears as a simple but realistic model for the alcohols carrying the main specificity of alcohols for example internal hydrogen bonds, and presenting primary and secondary OH groups. It is also a major by-product obtained during the production of biodiesel; thus the improvement in its conversion efficiency to value-added products not only has a positive effect on the economy, but also profits to sustainable development as an alternative energy resource [25].
The reactions leading to the current findings were carried out in a customized container for LAL as reported elsewhere [15, 24] Chemically pure gold clusters were synthesized by laser ablation of gold in water for which we performed high-resolution mass spectrometric analysis [15, 24]. Laser ablation of gold was done in pure water and the filtered product solution vaporized on a rotary vaporizer to concentrate the solution until the final volume was about 5 mL. Then 1 mL of glycerol was added, allowing for the reaction to proceed for 4 h at 70 ℃ prior to ESI-MS analysis. A similar procedure was also followed for methanol and ethanol. Exclusion experiments were also performed by carrying out the similar procedure without adding the gold cluster samples.
DFT calculations were performed using the B3PW91 hybrid exchange-correlation functional [26], which has been found to provide a good compromise in describing both gold clusters and organic molecules, as well as their interactions [27-32]. Triple-zeta level basis set with polarization functions [6-311 G(df, p)] was used for C, O, and H [33]. We used the scalar relativistic ECP valence double zeta plus polarization on all atoms (aug-cc-pVDZ-PP) for all gold atoms [34, 35]. In order to match the solvent employed in our experimental work, solvation effect was taken into consideration using the implicit continuum solvation model (with water as the solvent) [36]. As a self-consistent reaction field (SCRF) model, the integral equation formalism for polarizable continuum model (IEFPCM) was used. The transition states were located and characterized with a single imaginary frequency [37]. The differences of total electronic energies (ΔE) with corrections of zero point vibrations were used to elucidate the reaction coordinates. NBO analysis was performed using the related program implemented in the Gaussian 09 [38]. The second-order perturbative energy provides a measure of the overlap integral between the lone-pair orbital of the acceptor and the antibonding orbital of the donor [39], and it is beneficial in evaluating the contributions of a certain interaction to the molecule or complex stability.
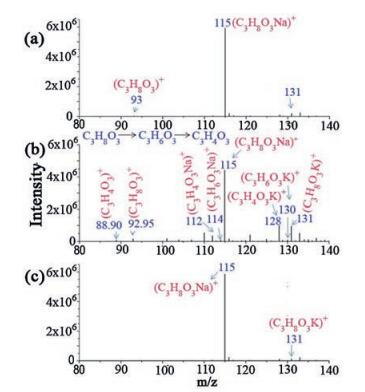
Fig. 1 shows the mass spectra of glycerol, and the products after reacting with the chemically pure gold clusters. As results, glycerol was found to be dehydrogenated to glyceraldehyde which underwent subsequent dehydrogenation to hydroxymethyl glyoxal with a yield of about 88%. An exclusion experiment was also performed by running the reaction in the absence of gold clusters as catalysts (Fig. 1c); where the dehydrogenation products were not observed. It is worth mentioning that, previously published studies also showed that gold and silver nanoparticles supported on hydrotalcite are efficient catalysts for the oxidantfree dehydrogenation of alcohols to carbonyl compounds with selectivity ranging between 47%–99% [20, 21]. These results are well consistent with this finding upon the small gold clusters. The small gold clusters were also found to be efficient models for a few other interesting catalytic reactions [40-43].

|
Download:
|
| Fig. 1. ESI-MS spectra of (a) pure glycerol, (b) glycerol after oxidant free reaction with the gold clusters, (c) reaction of glycerol in the absence of gold clusters as an exclusion experiment. | |
In order to verify the catalysis of such small gold clusters for the conversion of alcohols to carbonyl compounds, first-principles DFT calculations were employed to provide further insights into the reaction mechanism. We focused on Au5 as a representative in this study because such a small cluster is highly reproducible in our previous experiments [17]. In order to study the cluster size dependence on the interaction of alcohols with small gold clusters, site- and size- selective adsorptions of C3H8O3 on the small gold clusters are shown in Table S1 (Supporting information) where the binding energies and binding lengths are also provided. Among the small gold clusters that we studied, Au1 presents highest binding energy and shortest O-Au binding length; in comparison, Au2 gives the lowest binding energy. The structure of Au5 cluster is largely modified at the chemical adsorption of C3H8O3 (Fig. S1 in Supporting information), allowing the Au-O bond strength (i.e., binding energies) to vary with the adsorption sites. In particular, the adsorption of glycerol at the atom No. 3 of Au5 displays the shortest Au-O bond and highest binding energy. The C—O bond length in C3H8O3 (terminal C) is 1.46 Å, but after binding to Au5, it increases to 1.47 Å.
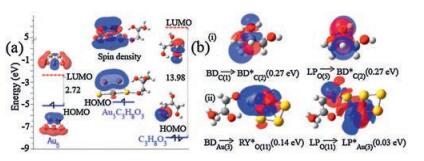
Fig. 2a displays the HOMOs, LUMOs, and their gaps for C3H8O3, Au5 and Au5C3H8O3. Au5 cluster has a low LUMO energy level that readily accepts an electron from the bonding HOMO of the Câ O bond in C3H8O3. The HOMO of Au5C3H8O3 is delocalized with the contribution from the Au atoms. Different from an individual C3H8O3 molecule where the contributions of C and O atoms to HOMO and LUMO can be seen, there is rare contribution of C3H8O3 in the HOMO of Au5C3H8O3 due to its low orbital energy. Fig. S2 (Supporting information) shows a comparison of the energy required to break C—H and O—H bonds in C3H8O3, comparing with the actual energy profile of the C—H and O—H bond stretching in the presence of the Au5 cluster catalyst. As is seen, typical case of the reduction of C—H and O—H bond breaking energies in C3H8O3 is dominated through the catalytic effect of Au5 cluster. It is also noted that the C—H bond-breaking energy is lower than that required to break the O—H bond. Further, NBO analysis has been performed to check possible interactions between the donor and acceptor in C3H8O3 along with the adsorbed complex. Typically, Fig. 2b(ⅰ) shows the interactions in C3H8O3. The dominant interactions are found to be mainly through the lone pair electrons on the O atom (LPO) and the antibonding lone pair (LPC*) of the C atom with a stabilization energy of 0.27 eV. In the adsorbed Au5-C3H8O3 complex (Fig. 2b (ⅱ)), Au5 interacts with the C3H8O3 mainly through the bonding orbital (BDAu) of Au and the Rydberg antibonding (RYO*) of the oxygen atom with stabilization energy of 0.14 eV. BDAu(3) is the higher-occupancy (0.81112 electrons) in comparison to RYO(9)* (0.00195 electrons) for C3H8O3. These interactions lead to donation of occupancy from the localized Lewis NBOs into the empty non-Lewis orbitals hence stabilizing the complexes through the Au–O bond. To further understand the electronic properties of Au5-C3H8O3 complex, a detailed Mulliken population analysis of the onsite atomic charges was performed for the lowest energy structures. Au5 bears -0.035 in Au5-C3H8O3 complex while C3H8O3 bears 0.96491, indicating that C3H8O3 is an electron donor allowing charge transfer from C3H8O3 to Au5. After the formation of stable Au-C3H8O3 complex, the spin density isosurface distributes largely on the molecular orbitals dominated by oxygen of C3H8O3 (Fig. 2a). That is, adsorption of C3H8O3 at the complimentary active sites of the cluster facilitates an electron transfer leading to a closure of electronic shell of the metal cluster.

|
Download:
|
| Fig. 2. (a) HOMOs, LUMOs, and HOMO-LUMO gaps of C3H8O3, Au5 and Au5C3H8O3. (b) Second order perturbation theory analysis of Fock matrix in NBO donor-acceptor interactions in, (ⅰ) C3H8O3 and (ⅱ) Au5-C3H8O3. The inserts indicate atom numbers, while BD (bonding orbital), BD* (antibonding orbital), LP (antibonding lone pair), RY (Rydberg orbital), RY* (Rydberg antibond) are molecular orbitals. Atoms in yellow, red, grey and white colour represent Au, O, C and H respectively. | |
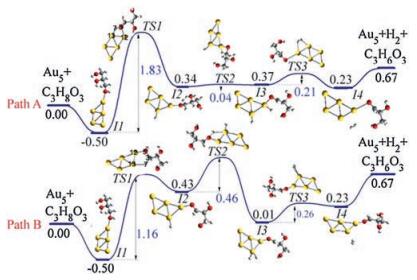
Fig. 3 plots the reaction coordinates involving elimination of two hydrogen atoms on C3H8O3 over Au5 to form C3H6O3. We have first considered the adsorption of the carbon atom to the Au cluster in the first transition state, whereby there is an insurmountable energy barrier of 15.62 eV and 17.12 eV (Fig. S3 in Supporting information). Next, we considered the transfer of H atom to the Au cluster, leading to C-H or O—H bond dissociation. As seen, in path A, the hydroxyl hydrogen atom is transferred to the Au5 cluster with a relatively high activation energy barrier of 1.83 eV in the first step; while hydrogen on the CH3 group of C3H8O3 is transferred to the Au5 cluster with an activation energy barrier, 1.16 eV in path B. To understand the electronic properties leading to the variation in the activation energy between path A and B (Fig. 3), we performed a detailed spin density and Mulliken population analysis of the onsite atomic charges for the lowest energy structures and transition states. Effective charges and spin densities are given in Table 1 and Fig. S4 (Supporting information).

|
Download:
|
| Fig. 3. The reaction coordinates of 'Au5 + C3H8O3' involving elimination of two hydrogen atoms on C3H8O3 over Au5 to form C3H6O3. (Path A) H on the OH group of C3H8O3 is transferred to the Au5 cluster in the first step. (Path B) H on the CH group of C3H8O3 is transferred to the Au5 cluster in the first step. Atoms in yellow, red, grey and white colour represent Au, O, C and H respectively. The inserts indicate atom numbers used in the discussion. Energies are in eV. | |
|
|
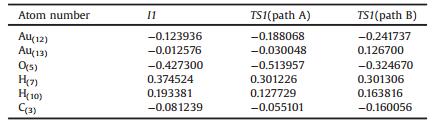
Table 1 Mulliken charges in the adsorption state (I1) and the first transition states (TS1) of 'Au5+C3H8O3'. |
{kind=link}
{kind=link}
{kind=link}
The values of spin densities and Mulliken charges on O(5) in TS1, in comparison to I1, suggest that O(5) acts as an electron donor in Path B (TS1) but as an electron acceptor in path A (TS1). On the other hand, Au(13) acts as an electron acceptor in path B (TS1) but as an electron donor in Path A (TS1). In the second step, H on the CH3 group is transferred to the Au cluster in path A; while H on the OH group is transferred to the Au cluster in path B, with single step energy barriers of 0.04 eV and 0.46 eV respectively. Note that, for the final dissociation step, there is a Gibbs free energy of 0.67 eV relative to the reaction energy of the initial reactants.
To gain further insights into dehydrogenation kinetics, we have then used the energetic span model to evaluate the energetic profile (Fig. S4). In Path A (Fig. 3), the first transition state, represented by the transfer of H from the OH group to the Au cluster (TS1), is the free-energy maximum of the whole catalytic cycle; while I1 is the free energy minimum. Thus, the turnover frequency determining intermediate (TDI) can be assigned to I1 while the turnover frequency determining transition state (TDTS) can be assigned toTS1. In Path B (Fig. 3), the second transition state, represented by the transfer of H from the OH group to the Au cluster (TS2), is the free-energy maximum of the whole catalytic cycle. Thus, the TDI can be assigned to I1 while the TDTS can be assigned to TS2. Because TDI comes before TDTS, the energy span can be calculated as ΔE = G(TDTS) - G(TDI). The corresponding turnover frequency (TOF) can be expressed as; TOF = exp-ΔE. It is seen that the calculated TOF for Path B (0.249) is higher than for Path A (0.160).
We also considered the co-adsorption of two C3H8O3 molecules, as seen in Fig. 4. Similar binding energies are observed in comparison to single molecule adsorption (i.e., 0.50 eV versus 0.51 eV). It is seen that the energy barrier for the transfer of H atom from the CH3 group to the Au cluster is lower (0.30 eV) than that for the 'Au5 + C3H8O3' path (1.16 eV). However, the H atom on the OH group is transferred to the Au cluster with a single step energy barrier of 0.8 eV.

|
Download:
|
| Fig. 4. The reaction coordinate of 'Au5 + (C3H8O3)2' involving elimination of two hydrogen atoms on (C3H8O3)2. Atoms in yellow, red, grey and white colour represent Au, O, C and H respectively. Energies are given in eV. | |
{kind=link}
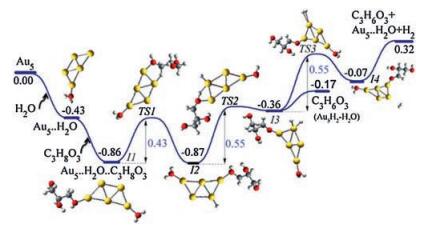
It is worth noting that chemically pure gold clusters synthesized by laser ablation of gold in water attach intact H2O molecules, hydrogen atoms and the Rydberg atoms (H3O) often bound at the active sites with donation of lone-pair electrons from the HOMO charge density of H2O to the LUMO (or LUMO + 1 for odd-electron systems) of the cluster, varying with the steric and electrostatic hindrance [15, 24]. Thus we considered the effect of the attached water molecules on the glycerol dehydrogenation pathway (Fig. 5). It is found that the cooperative adsorption of water and glycerol on the gold cluster gives a relatively higher binding energy (-0.86 eV). The first transition state energy, the subsequent single step energy barriers and the Gibbs free energy are also lower than that of the 'Au5 + C3H8O3' pathway (Fig. 3). Thus, water could function as a catalyst in the activation of the O—H and C—H bonds in glycerol [44].

|
Download:
|
| Fig. 5. The reaction coordinate of 'Au5 + H2O + C3H8O3' involving elimination of two hydrogen atoms on C3H8O3 over Au5H2O to form C3H6O3. Atoms in yellow, red, grey and white colour represent Au, O, C and H respectively. Energies are in eV. | |
{kind=link}
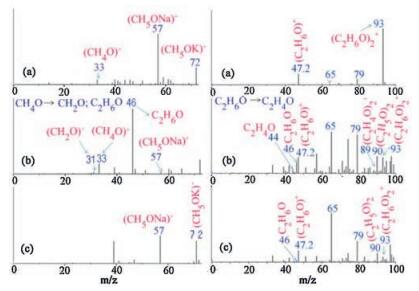
In comparison, methanol and ethanol were also found to dehydrogenate and form corresponding carbonyls (Fig. 6) although relatively low product yield of formaldehyde (3%) in comparison to subsequent side products (C2H6O and CH4). Also noted was that, the oxidant-free dehydrogenation to formaldehyde differs from the dehydrogenation of the other alcohols since formaldehyde is unstable under methanol dehydrogenation conditions and can decompose into CO and H2 [45]. The decomposition constant of formaldehyde to CO and H2 is higher than the equilibrium constant of the desired reaction by five orders of magnitude in the temperature range from 20 ℃ to 800 ℃ [45], and it is complicated with a variety of products over different zeolite catalysts [46].

|
Download:
|
| Fig. 6. (Left) ESI mass spectra of (a) pure methanol, (b) methanol after oxidant free reaction with the gold clusters, (c) reaction of methanol in the absence of gold clusters as an exclusion experiment. (Right) ESI mass spectra of (a) pure ethanol, (b) ethanol after oxidant free reaction with the gold clusters, (c) reaction of ethanol in the absence of gold clusters as an exclusion experiment. | |
{kind=link}
DFT calculations for the oxidant-free dehydrogenation of methanol and ethanol to form carbonyls over such small gold clusters have also been performed (Figs. S9 and S10 in Supporting information). In the proposed path where hydrogen on the OH group of CH3OH is transferred to the Au5 cluster, there is a relatively high activation energy barrier of 1.85 eV in the first step; while in the path for hydrogen on the CH3 group of CH3OH to be transferred to the Au5 cluster displays an activation energy barrier of 1.11 eV (Fig. S9), H on the CH3 group is transferred to the Au cluster in path A, while H on the OH group is transferred to the Au cluster in path B, with single step energy barriers 0.1 eV and 0.62 eV respectively. Note that, for the final dissociation step, a Gibbs free energy (relative to the reaction energy of the initial reactants) of 0.87 eV is observed. On the other hand, the first step in the CH3CH2OH dehydrogenation pathways also show a similar trend, whereby a higher activation energy barrier of 1.85 eV is observed in path A, in comparison to 1.11 eV in path B (Fig. S10). These results coincide with the previous work on Aln clusters in the gas phase [44], and also well consistent with the published studies on HAT process from alcohols to form carbonyls [20, 21, 47-51].
In summary, we have investigated the oxidant free conversion of glycerol to corresponding aldehydes over small gold clusters simply produced via a facile LAL method. On a basis of reaction kinetics calculations and NBO analysis, we provide insights into the mechanism of the C—O, C—H and O—H bond activation. Glycerol is found to dehydrogenate to form glyceraldehyde allowing subsequent dehydrogenation to produce hydroxymethyl glyoxal. Interestingly, lower transition-state energy barriers for the reaction path initiated by HAT from methene are observed in contrast to that from hydroxyl. Furthermore, it is revealed that coadsorbed water molecules, further lower the energy barriers to activate the O—H bond of glycerol. Similar mechanism is also found applicable to methanol and ethanol, verifying the feasible strategy of oxidant-free dehydrogenation catalyzed by small gold clusters via aqueous phase synthesis.
AcknowledgmentsZ. Luo acknowledges the National Thousand Youth Talents Program (No. Y3297B1261). This work was also financially supported by Key Research Program of Frontier Sciences (CAS, No. QYZDB-SSW-SLH024) and the National Natural Science Foundation of China (No. 21722308). A. Pembere acknowledges the support from the CAS/TWAS Presidential Fellowship Initiative for international students.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.12.019.
| [1] |
R. Uma, C. Crévisy, R. Grée, Chem. Rev. 103 (2003) 27-52. DOI:10.1021/cr0103165 |
| [2] |
P. Lorenzo-Luis, A. Romerosa, M. Serrano-Ruiz, ACS Catal. 2 (2012) 1079-1086. DOI:10.1021/cs300092d |
| [3] |
A.J. Mancuso, S.L. Huang, D. Swern, J. Org. Chem. 43 (1978) 2480-2482. DOI:10.1021/jo00406a041 |
| [4] |
R. Hajian, Z. Alghour, Chin. Chem. Lett. 28 (2017) 971-975. DOI:10.1016/j.cclet.2016.12.003 |
| [5] |
T.L. Liu, T.W. Ng, Y. Zhao, J. Am. Chem. Soc. 139 (2017) 3643-3646. DOI:10.1021/jacs.7b01096 |
| [6] |
A. Nakamura, A. Hamasaki, S. Goto, M. Utsunomiya, M. Tokunaga, Adv. Synth. Catal. 353 (2011) 973-984. DOI:10.1002/adsc.v353.6 |
| [7] |
P. Crochet, J. Díez, M.A. Fernández-Zúmel, J. Gimeno, Adv. Synth. Catal. 348 (2006) 93-100. |
| [8] |
C.A. Nunes, M.C. Guerreiro, J. Mol. Catal. A:Chem. 370 (2013) 145-151. DOI:10.1016/j.molcata.2013.01.006 |
| [9] |
E.A. Karlsson, T. Privalov, Chem.-Eur. J. 15 (2009) 1862-1869. DOI:10.1002/chem.v15:8 |
| [10] |
A. Gallo, C. Pirovano, P. Ferrini, et al., Appl. Catal. B-Environ. 121 (2012) 40-49. |
| [11] |
S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa, K. Tomishige, Appl. Catal. BEnviron. 111 (2012) 27-37. |
| [12] |
X.Y. Cheng, K.F. Li, Q.J. Wang, C.Y. Wang, T.K. Ying, Chin. Chem. Lett. 23 (2012) 801-804. DOI:10.1016/j.cclet.2012.05.019 |
| [13] |
T.E. Shubina, C. Hartnig, M.T. Koper, Phys. Chem. Chem. Phys. 6 (2004) 4215-4221. DOI:10.1039/b407669a |
| [14] |
H.Y. Su, M.M. Yang, X.H. Bao, W.X. Li, J. Phys. Chem. C 112 (2008) 17303-17310. DOI:10.1021/jp803400p |
| [15] |
D. Yuan, Z. Liu, J. Chen, J. Chem. Phys. 134 (2011) 054704. DOI:10.1063/1.3551617 |
| [16] |
H. Tsunoyama, H. Sakurai, Y. Negishi, T. Tsukuda, J. Am. Chem. Soc. 127 (2005) 9374-9375. DOI:10.1021/ja052161e |
| [17] |
A.M. Pembere, Z. Luo, Phys. Chem. Chem. Phys. 19 (2017) 6620-6625. DOI:10.1039/C6CP07941E |
| [18] |
G. Zhang, R. Wang, G. Li, Chin. Chem. Lett. 29 (2018) 687-693. DOI:10.1016/j.cclet.2018.01.043 |
| [19] |
P.G.N. Mertens, P. Vandezande, X. Ye, et al., Adv. Synth. Catal. 350 (2008) 1241-1247. DOI:10.1002/adsc.v350:9 |
| [20] |
W. Fang, Q. Zhang, J. Chen, W. Deng, Y. Wang, Chem. Comm. 46 (2010) 1547-1549. DOI:10.1039/b923047e |
| [21] |
T. Mitsudome, Y. Mikami, H. Funai, et al., Angew. Chem. Int. Ed. 120 (2008) 144-147. |
| [22] |
T. Mitsudome, Y. Mikami, K. Ebata, et al., Chem. Comm. (2008) 4804-4806. |
| [23] |
K.I. Shimizu, K. Sugino, K. Sawabe, A. Satsuma, Chem.-Eur. J. 15 (2009) 2341-2351. DOI:10.1002/chem.v15:10 |
| [24] |
A.M. Pembere, M. Yang, Z. Luo, Phys. Chem. Chem. Phys. 19 (2017) 25840-25845. DOI:10.1039/C7CP05324J |
| [25] |
A. Behr, J. Eilting, K. Irawadi, J. Leschinski, F. Lindner, Green. Chem. 10 (2008) 13-30. DOI:10.1039/B710561D |
| [26] |
K. Burke, J.P. Perdew, Y. Wang, Electronic Density Functional Theory, Springer, 1998, pp. 81-111.
|
| [27] |
A. Lyalin, T. Taketsugu, J. Phys. Chem. Lett. 1 (2010) 1752-1757. DOI:10.1021/jz100503j |
| [28] |
A. Lyalin, T. Taketsugu, J. Phys. Chem. C 113 (2009) 12930-12934. DOI:10.1021/jp903423j |
| [29] |
A. Lyalin, T. Taketsugu, J. Phys. Chem. C 114 (2010) 2484-2493. |
| [30] |
J. Li, X. Li, H.J. Zhai, L.S. Wang, Science 299 (2003) 864-867. DOI:10.1126/science.1079879 |
| [31] |
J.C. Idrobo, W. Walkosz, S.F. Yip, et al., Phys. Rev. B 76 (2007) 205422. DOI:10.1103/PhysRevB.76.205422 |
| [32] |
L. Xiao, B. Tollberg, X. Hu, L. Wang, J. Chem. Phys. 124 (2006) 114309. DOI:10.1063/1.2179419 |
| [33] |
J.D. Dill, J.A. Pople, J. Chem. Phys. 62 (1975) 2921-2923. DOI:10.1063/1.430801 |
| [34] |
P. Pyykkö, Angew. Chem. Int. Ed. 43 (2004) 4412-4456. |
| [35] |
D. Figgen, K.A. Peterson, M. Dolg, H. Stoll, J. Chem. Phys. 130 (2009) 164108. DOI:10.1063/1.3119665 |
| [36] |
S. Miertus, J. Tomasi, Chem. Phys. 65 (1982) 239-245. DOI:10.1016/0301-0104(82)85072-6 |
| [37] |
C. Gonzalez, H.B. Schlegel, J. Chem. Phys. 90 (1989) 2154-2161. DOI:10.1063/1.456010 |
| [38] |
E.D. Glendening, A. Reed, J. Carpenter, F. Weinhold, NBO Version 3.1, University of Wisconsin, Madison, 1998.
|
| [39] |
E.D. Glendening, C.R. Landis, F. Weinhold, WIRES Comput. Mol. Sci. 2 (2012) 1-42. DOI:10.1002/wcms.51 |
| [40] |
A. Nijamudheen, D. Jose, A. Datta, J. Phys. Chem. C 115 (2010) 2187-2195. |
| [41] |
C.R. Chang, Y.G. Wang, J. Li, Nano Res. 4 (2011) 131-142. DOI:10.1007/s12274-010-0083-8 |
| [42] |
S.M. Lang, T.M. Bernhardt, Faraday Disc 152 (2011) 337-351. DOI:10.1039/c1fd00025j |
| [43] |
S.M. Lang, T.M. Bernhardt, R.N. Barnett, U. Landman, Angew. Chem. Int. Ed. 49 (2010) 980-983. DOI:10.1002/anie.200905643 |
| [44] |
Z. Luo, J.C. Smith, W.H. Woodward, A.W. Castleman, J. Phys. Chem. Lett. 3 (2012) 3818-3821. DOI:10.1021/jz301830v |
| [45] |
N.Y. Usachev, I. Krukovskii, S. Kanaev, Petrol. Chem. 44 (2004) 379-394. |
| [46] |
P. Tian, Y. Wei, M. Ye, Z. Liu, ACS Catal. 5 (2015) 1922-1938. DOI:10.1021/acscatal.5b00007 |
| [47] |
L. Deng, T. Ziegler, Organometallics 16 (1997) 716-724. DOI:10.1021/om9606606 |
| [48] |
H. Li, C. Mazet, Acc. Chem. Res. 49 (2016) 1232-1241. DOI:10.1021/acs.accounts.6b00144 |
| [49] |
J. Mobley, M. Crocker, RSC Adv. 5 (2015) 65780-65797. DOI:10.1039/C5RA11254K |
| [50] |
L. Cheng, J. Li, Q. Zhang, L. Ma, J. Yang, Dalton Trans. 44 (2015) 7395-7403. DOI:10.1039/C4DT03051F |
| [51] |
J.M. Hoover, B.L. Ryland, S.S. Stahl, J. Am. Chem. Soc. 135 (2013) 2357-2367. DOI:10.1021/ja3117203 |