 2019, Vol. 30
2019, Vol. 30 


b School of Chemistry and Chemical Engineering, Tianjin University of Technology, Tianjin 300384, China;
c Institute for New Energy Materials and Low Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China
The molecular ORR is fundamental importance in both biological and industrial processes [1-3]. In the industrial process, the efficient ORR electrocatalysts are essential in various fields of energy, such as fuel cells, metal–air cells, solar cells and airbreathing cathodes in industrial electrocatalytic processes [4-6]. Traditionally, Pt-based electrocatalysts have been widely utilized to efficiently catalyze ORR in electrochemical fuel cells because of their high activity in acidic, neutral and alkaline solutions [7-10]. Nevertheless, it still suffers from multiple drawbacks, such as high cost, methanol crossover, and CO poisoning effects, which enormously restrict their practical applications. Hence, it is urgently needed to explore and develop high-activity, robust, and earth-abundant alternatives to achieve efficient ORR. Up to now, a great deal of efforts have been made to develop high performance and durable non-noble metal ORR catalysts, such as non-noble metal oxides, nitrides, and phosphides, mixed transition metals and/or nitrogen co-doped carbon materials and single atom catalysts [11-21].
Metal-organic frameworks (MOFs), a kind of highly ordered porous materials with easily tunable components and welldefined structures [22-27], are particularly attractive as ideal precursors to install functionality via the calcination process [28-32]. The MOF-derivatives have been widely recognized as efficient functional materials for activating small molecules, such as H2O, CO2 and O2 [33-35]. The combination of MOFs with carbon materials has been considered as an efficient strategy to functionalize the carbon-based materials. In this field, graphene, carbon fiber and ordered mesoporous carbon are usually used as the substrates to support MOFs. The multiple components of MOFs, such as transition metals, N, P and S can be readily introduced into the carbon substrates in the pyrolysis process to greatly improve their performance [36-45]. In this field, Wang et al. explored a facile strategy for preparing ORR electrocatalyst by pyrolysis of CoZIF/CC [37], where a very cheap industrial carbon black Vulcan XC72 was used as the support. As well known, industrial carbon black with porous structure and high conductivity is much cheaper compared to graphene and carbon nanotubes [46, 47]. Also, cobalt is one of the most earth-abundant metal candidates for preparing ORR electrocatalysts. Thus, combining Co-MOFs with carbon black may represent a promising way to endow CC with excellent ORR catalytic activity. However, the systematical study of wellcontrolled calcination of Co-MOF/CC and the affecting factors with versatility of MOFs has not yet been widely realized.
Herein, two carboxylate ligands with different nitrogen contents were used for in situ coating different Co-MOFs on the surface of CC, resulting in two Co-MOF/CC composites, MOF-1/CC and MOF-2/CC. The structure of MOFs Co2(INA)4·DMF (1) and {[Co2(TPI) (H2O)2(OH)]·DMF} (2) was determined by single crystal X-ray diffraction analysis. Further, the composites MOF-1/CC and MOF-2/CC were calcinated into Cal-MOF-1/CC (3) and Cal-MOF-2/CC (4), where the CC was doped by trace amount of cobalt oxides and nitrogen element. The ORR performance of 3 and 4 was much enhanced compared to that of pristine CC and 4 was even exceeded that of commercial Pt/C. Also, this functionalized CC shows long endurance and can resist methanol poisoning in 0.1 mol/L KOH aqueous solution.Detailed study on the structure and component of MOFs and Cal-MOF/CC demonstrates that the carboxylate ligands with different nitrogen contents play important role in constructing different crystal structures, and smartly influence the activityof the composites. As a result, the catalytic activity of Co3O4/CoO/Co codoped CC in 4 was much higher than that of CoO-doped CC in 3.
The synthetic process of 1–4 is schematically depicted in Fig. S1 (Supporting information). For in situ coating these two MOFs on CC, the commercially obtained Vulcan XC72 was directly introduced into the ethanol solution of Co(NO3)2-6H2O to support cobalt cations, resulting in cobalt-containing CC (Co/CC). The obtained Co/CC was further used as raw material to replace Co(NO3)2-6H2O under similar solvothermal condition with the synthesis of single crystal of 1 and 2. Finally, the obtained MOF-1/CC and MOF-2/CC were further calcinated in a tubular furnace at 800 ℃ for 6 h under N2 flow, resulting in CC coated with cobalt oxide nanoparticles in 3 and 4. For a comparison, the Co/CC was also calcinated under the same condition to prepare Cal-Co/CC.
In the synthesis of the MOFs, the HINA and H3TPI ligands were used to synthesize 1 and 2 under solvothermal conditions, respectively. Single crystal X-ray diffraction analysis reveals that compound 1 crystallizes in the monoclinic space group P2(1), exhibiting a 3D framework with 1D channels along the a-axis. The asymmetric unit contains two Co(Ⅱ) ions, four INA ligands and a DMF molecule. The Co(Ⅱ) centers are both in an octahedral hexacoordinated environment completed by four carboxylate oxygen atoms and two pyridine nitrogenatoms. In an asymmetric unit, two Co(Ⅱ) centers are fused together by two carboxylate ligands with the Co-μ2–COO-Co linking mode. The Co–O and Co–N distances are in the range of 2.069(4)–2.15(4) Å and 2.129(4)–2.161(4) Å, respectively. As shown in Fig. 1A, each INA ligand linked with three metal ions via the pyridine N atom and carboxylate oxygen atoms, and each Co2+ ion connected with six INA ligands, generating a (3, 6)-connected 3D network with 1D channel along the a-axis (Figs. 1C and D). Compound 2 crystallizes in the monoclinic space group C2/c exhibiting a 3D framework with a 1D tubular channel. In one asymmetric unit, three Co(Ⅱ) ions were fused together by a OH group and six N atoms from three TPI ligands into a triangular unit, which was further expended into 3D porous framework (Fig. 1B and Fig. S2 in Supporting information). This structure has been studied in detail by Wang et al. [48].

|
Download:
|
| Fig. 1. Coordination modes of (A) Co(Ⅱ) ions and INA in 1, and (B) Co(Ⅱ) ions and TPI in 2. (C, D) View of 3D porous framework of 1 along a-axis. All hydrogen atoms are omitted for clarity. | |
{kind=link}
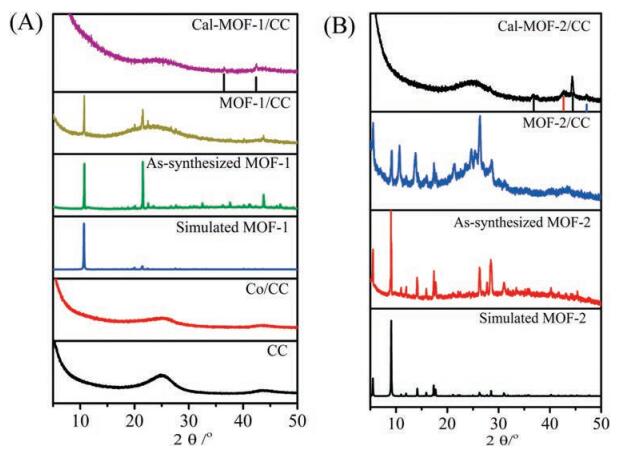
PXRD measurement proves the successful synthesis of Co-MOFs on CC (Fig. 2), which was further transferred into Co oxide nanoparticles. As shown in Fig. 2, the XRD peaks of these Co-MOFs and Co-MOFs on CC are similar to the simulated patterns from single crystal data, indicating a purity of the bulk samples, and the successful synthesis of Co-MOFs on CC. After calcination, the diffraction peaks of the MOFs disappeared. For 3, only two peaks in consistent with (111) and (200) facets of CoO (JCPDS card No. 43-1004) were observed at 36.5° and 42.4° (Fig. 2A). In the XRD of 4, four weak peaks related to (311) and (400) facets of Co3O4 (JCPDS card No. 43-1003), (200) facets of CoO (JCPDS card No. 43-1004) and (101) facets of Co (JCPDS card No. 15-0727) were detected at 36.845°, 44.808°, 42.4° and 47.568°, respectively (Fig. 2B).

|
Download:
|
| Fig. 2. (A) XRD patterns of CC (black), Co/CC (red), simulated 1 (blue), as synthesized 1 (green), MOF-1/CC (yellow-green), and 3 (purple). (B) XRD patterns of simulated 2 (black), as synthesized 2 (red), MOF-2/CC (blue), and 4 (black). | |
{kind=link}
To further analyze the morphology and composition, highresolution transmission electron microscopy (HRTEM) was performed on the as-synthesized 3 and 4. As shown in Fig. 3 and Figs. S3, S4 (Supporting information), the carbon matrix of 3 and 4 maintains the morphology of pristine CC (Fig. S5 in Supporting information), while the nanoparticles randomly distributed on the carbon matrix. HRTEM images of 3 show the crystalline nanoparticles were embedded in the carbon matrix, and the crystalline lattice spacings of these nanoparticles are 0.245 nm and 0.213 nm (Fig. S3B), corresponding to the (111) and (200) crystal orientation of CoO. As shown in Fig. 3B, the crystal lattice spacings on the crystalline nanoparticles in 4 are 0.466 nm, 0.243 nm, 0.213 nm and 0.191 nm. The first two lattice spacings correspond to the (111) and (311) crystal orientation of Co3O4, the first two lattice spacings can be attributed to the (200) crystal orientation of CoO and (101) crystal orientation of Co, respectively. Energy-dispersive X-ray spectroscopy (EDS) elemental mapping images also confirmed that the majority of Co and O elements distribute in the crystalline cobalt oxide nanoparticles on the carbon matrix (Figs. S3C–G and Figs. 3C–G). EDS study indicated the existence of trace amount of Co (Fig. S6 in Supporting information), which is consistent with inductively coupled plasma mass spectrometry (ICP-MS) results. The loading amount of Co content in 3 and 4 was determined to be 3.3 wt% and 4.2 wt%, respectively.

|
Download:
|
| Fig. 3. (A) TEM image of 4. (B) HRTEM image of 4 showing the crystal orientation of Co3O4, CoO and Co. (C–G) EDS elemental mapping images of 4. | |
{kind=link}
All the above results confirmed that trace amount of cobalt oxide nanoparticles were anchored on low-cost CC. However, only peaks of O and C were detected from the XPS spectra, no obvious peaks of other doping elements were clearly observed (Fig. S7A in Supporting information) [49]. For the C 1s, the XPS spectra of CC, 3 and 4 all exhibit three peaks arising from C—C (284.8 eV), C–O (285.5 eV) and O—C=O (291.6 eV) bonds (Supporting information). From a total view, no obvious changes were observed from the XPS spectra after loading the nanoparticles compared to the pristine CC, only the peak of O 1s in 3 and 4 become a little stronger than that of pristine CC, indicating some more oxygen element was introduced into the C matrix with loading the cobalt oxides. Furthermore, no peak shifts and new peak formation in the FT-IR spectra of 3 and 4 could be detected in comparison with that of pristine CC (Fig. S9A in Supporting information). In the Raman spectra, the D-band centred at ≈ 1360 cm-1 of sp3 carbons and Gband centred at ≈1580 cm-1 of all sp2 carbons were both detected with a similar appearance [50, 51], and the proportions of the peak area of D-band to that of G-band in CC (1.00), 3 (1.05) and 4 (1.05) are almost the same within the margin of error in the peak fitting (Fig. S9B in Supporting information). Therefore, it can be deduced that there is no obvious structural change the carbon matrix in 3 and 4 by loading trace amount of cobalt oxides.
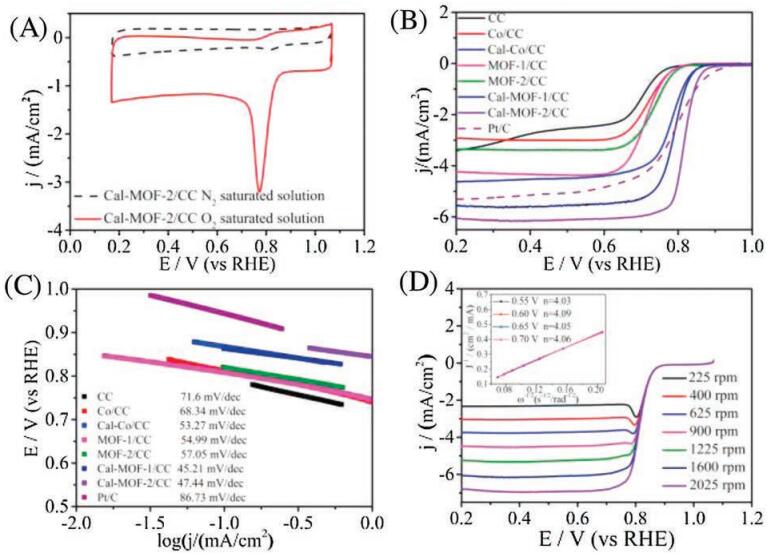
The electrocatalytic ORR activity of 3 and 4 was investigated in 0.1 mol/L KOH solution with 3 or 4 decorated electrode, Ag/AgCl (saturated KCl) electrode and a carbon rod as working electrode, reference electrode and counter electrode, respectively. Firstly, the cyclic voltammetry (CV) curves of 3 and 4 were examined in N2 and O2 saturated 0.1 mol/L KOH aqueous solution. As depicted in Fig. S10 (Supporting information) and Fig. 4A, an obvious cathodic peak was detected in the O2-saturated electrolyte, whereas no obvious reduction peak was found in the N2-saturated solution. The ORR performance of 3, 4, 20% Pt/C and some other comparative catalysts was further examined by the linear scanning voltammetry (LSV). As shown in Figs. 4B and C, the activity of Co/CC obtained by immersing CC into Co(NO3)2 solution for 12 h without further treatment was almost the same as that of original CC (jL 2.8 mA/cm2 and E 1/2 0.69 V), showing similar LSV and Tafel curves. After coating 1 and 2 on CC (denoted as MOF-1/CC and MOF-2/CC), their activity was obviously enhanced with larger jL (4.2 mA/cm2 for MOF-1/CC and 3.3 mA/cm2 for MOF-2/CC) and E1/2 (0.71 V for MOF-1/CC and 0.72 V for MOF-2/CC), and their Tafel slopes are 54.99 and 57.05 mV/dec, respectively. However, the MOF particles cannot be obviously detected in MOF-1/CC and MOF-2/CC from the TEM images (Figs. S11 and S12 in Supporting information). This situation has been usually observed in the MOF/carbon composites. After calcinations, the jL and E1/2 of Cal-Co/CC are 5.64 mA/cm2 and 0.80 V, 6.04 mA/cm2 and 0.82 V for 3 and 4, respectively. In addition, the Tafel slopes of 3 and 4 are 45.21 and 47.44 mV/dec, respectively. Typically, when Tafel slope is closed to 40 mV/dec, the order of reaction is -1 in OH-. When the Tafel slope is close to 60 or 120 mV/dec, there are different ratedetermining steps in the reaction mechanism [37, 52]. The Tafel slope of 3 and 4 are smaller than Pt/C (86.73 mV/dec), indicating faster reaction kinetics for ORR with 3 and 4 as the catalysts. These Tafel slope values are not typical, suggesting a complicated mechanism in the reaction [37]. They both showed much improved ORR performance compared with that of pristine CC, MOF/CC, and 4 was even better than that of commercial 20% Pt/C. In addition, the ORR activity of 4 is much superior to that of 3, showing the best activity in line with the most positive half-wave potential E1/2 as well as higher diffusion-limiting current density jL among these comparative catalysts.

|
Download:
|
| Fig. 4. (A) CV plots of 4 in N2-saturated and O2-saturated 0.1 mol/L KOH aqueous solution at a scan rate of 10 mV/s. (B) LSV curves and (C) Tafel slopes of 3, 4 and other comparative catalysts at 1600 rpm. (D) LSV curves of 4 at different rotating speeds of RDEI, showing the K–L plots at various potentials. | |
{kind=link}
To investigate the electron transfer mechanism, the LSV curves were recorded on the RDE with different rotating speeds, ranging from 225 rpm to 2025 rpm. The electron transfer number (n) and kinetic current density (jK) of 3 and 4 were calculated by the Koutecky–Levich (K–L) equation [53]. As shown in Fig. S13 (Supporting information) and Fig. 4D, the jL of 3 and 4 both increased with the increase of rotating speeds owing to the decrease of electron and ion diffusion distance. In addition, the corresponding K–L plots obtained from LSV curves at different potentials of 3 and 4 were almost coincide (inset of Figs. S13 and Fig. 4D), indicating similar electron transfer numbers, which were both calculated to be approximately 4.0. These results confirmed that 3 and 4 underwent an ideal one-step four-electron catalytic process (O2 + 2H2O + 4e- = 4OH-), and could act as efficient catalysts to directly reduce O2 to H2O. Comparing the intercepts of K–L plots at -0.75 V for 3 and 4, the jK of them were about 15.25 mA/cm2 and 15.86 mA/cm2 (Fig. S14 in Supporting information), indicating a high kinetic activity of 3 and 4.
Furthermore, the durability of 3 and 4 was evaluated by CV measurement between the potential range 0.4 V and 1.0 V with a scan rate of 100 mV/s in O2-saturated 0.1 mol/L KOH electrolyte solution. After 1000 cycles, no obvious change of E1/2 could be detected (Figs. S15A and B in Supporting information), demonstrating high stability of 3 and 4 in the ORR process. To examine their tolerance to methanol crossover effect, the ORR was performed with 3 and 4 as the catalysts in O2-stratured 0.1 mol/L KOH aqueous solution and O2-stratured 0.1 mol/L KOH aqueous solution containing 1.0 mol/L methanol, respectively. After the injection of 1.0 mol/L methanol into the solution, the current density and E1/2 of 3 and 4 did not change significantly (Figs. S15C and D in Supporting information). Also, the LSV curves of 4 before and after 1000 cycles in 0.1 mol/L KOHsolutionwith 1.0 mol/L methanol(Fig. S15D), show similar results to that of 4 in 0.1 mol/L KOH solution, indicating its excellent tolerance to methanol crossover.
Different activity of 3 and 4 can be attributed to the formation of different MOFs with different structures, linking ligands and N contents in MOF/CC precursors. In these two MOFs, different linking ligands directly lead to different aggregation mode and coordination environment of Co2+ centers. In MOF-1, mononuclear Co center was completely isolated by organic linking ligands, and trinuclear cobalt cluster formed in MOF-2. In the calcination process, these two MOFs with different structure and different CoCo distances lead to different oxides and N contents in the calcined products, which may play important role in improving the activity of these carbon materials [36-39]. Also, element analysis was further performed, confirming that the N content of 4 (0.37%) is higher than that of 3 (0.2%). In addition, surface area of these composites is also an important factor to influence their catalytic activity. Electrochemical active surface area (ECSA) of 3 and 4 were detected, which can be represented by double-layer capacitance (CdI) obtained by measuring CV curves at different scan rates. As shown in Fig. S16 (Supporting information), 4 has a higher CdI value of 31.25 mF/cm2, about 3.1 times higher than that of 3 (10.15 mF/cm2). The N2 adsorption/desorption isotherms were also performed on CC, 3 and 4. The BET surface area of 4 was 1075.3 m2/g, about 6.3 times than that of 3 (171.19 m2/g), indicating that 4 possesses of higher surface area to accommodate more active sites. Overall, all the above results support that the MOFs with different structures, linking ligands and N contents strongly affect the components, surface area, and active site distribution of the functionalized carbon materials to further improve their ORR performance.
In summary, two efficient ORR electrocatalysts 3 and 4 were successfully prepared by a simple absorption-reaction method by directly coating two kinds of MOFs on CC. The resulting MOF/CC composites were used as precursors to afford trace amount cobalt oxide on low-cost CC by a subsequent carbonization process. The final composites of 3 and 4 both exhibit excellent electrocatalytic ORR activity. Also, they both exhibit good durability and excellent tolerance to methanol crossover during the ORR. In light of the tremendous diversity and tailorability of MOFs, the linking ligands with different structure and nitrogen contents smartly influence structure of the MOFs to further affect the components, surface area, and active site distribution of the functionalized carbon materials. Thus, the catalytic activity of Co3O4/CoO/Co co-doped CC in 4 was much enhanced compared to that of CoO-doped CC in 3. This work may open an avenue in the rational design and synthesis of high-efficiency, low-cost, and robust ORR electrocatalysts with different MOFs.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21722104, 21671032), Natural Science Foundation of Tianjin City of China (Nos. 18JCJQJC47700, 17JCQNJC05100), Research Foundation of Thirteenth Five Years of Jilin Educational Committee (No. [2015]0056/JJKH20170605KJ), the Scientific Research Foundation for the Returned Overseas Scholars.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.02.021.
| [1] |
K.P. Gong, F. Du, Z.H. Xia, M. Durstock, L.M. Dai, Science 323 (2009) 760-764. DOI:10.1126/science.1168049 |
| [2] |
Q. Bai, F.C. Shen, S.L. Li, et al., Small (2018) 1800049. |
| [3] |
B. Šljukić, C.E. Banks, R.G. Compton, J. Iran. Chem. Soc. 2 (2005) 1-25. DOI:10.1007/BF03245775 |
| [4] |
B.C.H. Steele, A. Heinzel, Nature 414 (2001) 345-352. DOI:10.1038/35104620 |
| [5] |
J.S. Lee, G. Nam, J. Sun, et al., Adv. Energy Mater. 6 (2016) 1601052. DOI:10.1002/aenm.v6.22 |
| [6] |
C. Tang, B. Wang, H.F. Wang, Q. Zhang, Adv. Mater. 29 (2017) 1703185. DOI:10.1002/adma.v29.37 |
| [7] |
D.P. He, Z.K. Kou, S.C. Mu, et al., Carbon 66 (2014) 312-319. DOI:10.1016/j.carbon.2013.09.005 |
| [8] |
H.F. Lv, S.C. Mu, et al., Nanoscale 6 (2014) 5063-5074. DOI:10.1039/C4NR00402G |
| [9] |
C. Meng, T. Ling, T.Y. Ma, et al., Adv. Mater. 29 (2017) 1604607. DOI:10.1002/adma.201604607 |
| [10] |
K. Jiang, P.T. Wang, S.J. Guo, et al., Angew. Chem. Int. Ed. 55 (2016) 9030-9035. DOI:10.1002/anie.201603022 |
| [11] |
R. Bashyam, P. Zelenay, Nature 443 (2006) 63-66. DOI:10.1038/nature05118 |
| [12] |
Z.W. Chen, D. Higgins, A.P. Yu, et al., Energy Environ. Sci. 4 (2011) 3167-3192. DOI:10.1039/c0ee00558d |
| [13] |
A. Miura, C. Rosero-Navarro, Y. Masubuchi, et al., Angew. Chem. Int. Ed. 55 (2016) 7963-7967. DOI:10.1002/anie.201601568 |
| [14] |
J.L. Shui, M. Wang, F. Du, L.M. Dai, Sci. Adv. 1 (2015) e1400129. DOI:10.1126/sciadv.1400129 |
| [15] |
K. Iwase, T. Yoshioka, S. Nakanishi, K. Hashimoto, K. Kamiya, Angew. Chem. Int. Ed. 54 (2015) 11068-11072. DOI:10.1002/anie.201503637 |
| [16] |
H.J. Tang, H.J. Yin, J.Y. Wang, et al., Angew. Chem. Int. Ed. 52 (2013) 5585-5589. DOI:10.1002/anie.201300711 |
| [17] |
G. Wu, K.L. More, C.M. Johnston, P. Zelenay, Science 332 (2011) 443-447. DOI:10.1126/science.1200832 |
| [18] |
W.H. Niu, L.G. Li, X.J. Liu, et al., J. Am. Chem. Soc. 137 (2015) 5555-5562. DOI:10.1021/jacs.5b02027 |
| [19] |
J. Liang, R.F. Zhou, X.M. Chen, Y.H. Tang, S.Z. Qiao, Adv. Mater. 26 (2014) 6074-6079. DOI:10.1002/adma.201401848 |
| [20] |
M. Lefevre, E. Proietti, F. Jaouen, J.P. Dodelet, Science 324 (2009) 71-74. DOI:10.1126/science.1170051 |
| [21] |
H.W. Liang, W. Wei, Z.S. Wu, X.L. Feng, K. Müllen, J. Am. Chem. Soc. 135 (2013) 16002-16005. DOI:10.1021/ja407552k |
| [22] |
Q. Yang, Q. Xu, H.L. Jiang, Chem. Soc. Rev. 46 (2017) 4774-4808. DOI:10.1039/C6CS00724D |
| [23] |
K. Shen, X. Chen, J. Chen, Y.W. Li, ACS Catal. 6 (2016) 5887-5903. DOI:10.1021/acscatal.6b01222 |
| [24] |
L.L. Wu, Q.S. Wang, J. Li, et al., Small (2018) 1704035. |
| [25] |
H.C. Zhou, J.R. Long, O.M. Yaghi, Chem. Rev. 112 (2012) 673-674. DOI:10.1021/cr300014x |
| [26] |
Z.M. Zhang, T. Zhang, C. Wang, et al., J. Am. Chem. Soc. 137 (2015) 3197-3200. DOI:10.1021/jacs.5b00075 |
| [27] |
Q. Lan, Z.M. Zhang, C. Qin, et al., Chem.-Eur. J. 22 (2016) 15513-15520. DOI:10.1002/chem.v22.43 |
| [28] |
Q.L. Zhu, W. Xia, T. Akita, R.Q. Zou, Q. Xu, Adv. Mater. 28 (2016) 6391-6398. DOI:10.1002/adma.201600979 |
| [29] |
Y.J. Chen, S.F. Ji, Y.G. Wang, et al., Angew. Chem. Int. Ed. 56 (2017) 6937-6941. DOI:10.1002/anie.201702473 |
| [30] |
C.M. Wang, X.Y. Jin, T. Yao, et al., J. Am. Chem. Soc. 139 (2017) 8078-8081. DOI:10.1021/jacs.7b02736 |
| [31] |
P.Q. Yin, T. Yao, Y.E. Wu, et al., Angew. Chem. Int. Ed. 55 (2016) 10800-10805. DOI:10.1002/anie.201604802 |
| [32] |
H.X. Zhong, J. Wang, Y.W. Zhang, et al., Angew. Chem. Int. Ed. 53 (2014) 14235-14239. DOI:10.1002/anie.v53.51 |
| [33] |
J.D. Xiao, L.L. Han, J. Luo, S.H. Yu, H.L. Jiang, Angew. Chem. Int. Ed. 57 (2018) 1103-1107. DOI:10.1002/anie.201711725 |
| [34] |
L. Jiao, G. Wan, R. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 8525-8529. DOI:10.1002/anie.201803262 |
| [35] |
L.M. Cao, Y.W. Hu, S.F. Tang, et al., Adv. Sci. 5 (2018) 1800949. DOI:10.1002/advs.v5.10 |
| [36] |
M.D. Zhang, Q.B. Dai, M.D. Chen, L.M. Dai, Adv. Mater. 30 (2018) 1705431. DOI:10.1002/adma.v30.10 |
| [37] |
B. Ni, C. Ouyang, X.B. Xu, J. Zhuang, X. Wang, Adv. Mater. 29 (2017) 1701354. DOI:10.1002/adma.v29.27 |
| [38] |
I.S. Amiinu, Z.H. Pu, X.B. Liu, et al., Adv. Funct. Mater. 27 (2017) 1702300. DOI:10.1002/adfm.v27.44 |
| [39] |
I.S. Amiinu, X.B. Liu, Z.H. Pu, et al., Adv. Funct. Mater. 28 (2018) 1704638. DOI:10.1002/adfm.v28.5 |
| [40] |
Q.R. Liang, H.H. Jin, Z. Wang, et al., Nano Energy 57 (2019) 746-752. DOI:10.1016/j.nanoen.2018.12.060 |
| [41] |
Z.H. Wang, H.H. Jin, T. Meng, et al., Adv. Funct. Mater. 28 (2018) 1802596. DOI:10.1002/adfm.v28.39 |
| [42] |
B.L. Chen, G.P. Ma, Y.Q. Zhu, Y.D. Xia, Sci. Rep. 7 (2017) 5266. DOI:10.1038/s41598-017-05636-y |
| [43] |
G. Huang, Y.Z. Chen, H.L. Jiang, Acta Chim. Sin. 74 (2016) 113-129. DOI:10.6023/A15080547 |
| [44] |
Y.Z. Chen, R. Zhang, L. Jiao, H.L. Jiang, Coord. Chem. Rev. 362 (2018) 1-23. DOI:10.1016/j.ccr.2018.02.008 |
| [45] |
H.Y. Yang, W.H. Cui, Y.Z. Han, B. Wang, Chin. Chem. Lett. 29 (2018) 842-844. DOI:10.1016/j.cclet.2017.09.024 |
| [46] |
B. Ni, X. Wang, Chem. Sci. 6 (2015) 3572-3576. DOI:10.1039/C5SC00836K |
| [47] |
H. Liu, H. Li, X. Wang, Small 12 (2016) 2969-2974. DOI:10.1002/smll.201600345 |
| [48] |
H.H. Wang, L. Hou, Y.Z. Li, et al., ACS Appl. Mater. Interfaces 9 (2017) 17969-17976. DOI:10.1021/acsami.7b03835 |
| [49] |
Y.J. Tang, C.H. Liu, W. Huang, et al., ACS Appl. Mater. Interfaces 9 (2017) 16977-16985. DOI:10.1021/acsami.7b01096 |
| [50] |
F. Tuinstra, J.L. Koenig, J. Chem. Phys. 53 (1970) 1126-1130. DOI:10.1063/1.1674108 |
| [51] |
M.S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus, R. Saitoet, Nano Lett. 10 (2010) 751-758. DOI:10.1021/nl904286r |
| [52] |
J.H. Zagal, M.T.M. Koper, Angew. Chem. Int. Ed. 55 (2016) 14510-14521. DOI:10.1002/anie.201604311 |
| [53] |
C. Liu, J. Wang, J.S. Li, et al., J. Mater. Chem. A 5 (2017) 1211-1220. DOI:10.1039/C6TA09193H |