 2019, Vol. 30
2019, Vol. 30 


b School of Pharmacy, Anhui University of Chinese Medicine, Anhui Academy of Chinese Medicine, Hefei 30012, China;
c Department of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK
Photocatalytic water splitting has been extensively studied as an ideal method to produce hydrogen since the report of the Honda-Fujishima effect of water splitting using a TiO2 electrode [1]. It consists of photoexcitation of semiconductor photocatalyst to generate electrons and holes in the bulk, transfer of photoexcited carriers from the bulk to the surface, and photoexcited electrons-participated water reduction to H2 and photoexcited holes-participated water oxidation to O2 on the surface [2-4]. Among various semiconductor photocatalysts for photocatalytic water splitting, TiO2 is considered as a potential commercial photocatalyst due to the favorable electronic energy band structure and high photo-chemical stability [5-7]. However, TiO2 photocatalyst suffers from the wide band gap of around 3.2 eV [8-10] and rapid recombination of photoexcited electronhole pairs [5, 6]. Great effort has been thus devoted to suppressing the recombination of photoexcited electron-hole pairs to increase the efficiency of TiO2 in photocatalytic water splitting, and the cocatalyst strategy is very effective. Noble metals (Pt, Pd, Au and Ag) and transitional metal oxides have been demonstrated as efficient co-catalysts for TiO2 to suppress the charge recombination via the transfer of photoexcited electrons and holes from TiO2 to the cocatalyst, respectively [11-20]. Recently space-separated noble metal and transitional metal oxide co-catalysts have been successfully loaded on TiO2 to allow simultaneous transfer of photoexcited electrons and holes from TiO2 respectively to the noble metal and transitional metal oxide co-catalysts [11-13].
Utilizing photoexcited electrons to reduce metal cations, photodeposition emerges as a convenient method to load metal particles on semiconductors [21-28]. Photodeposition was also successfully used for a simultaneous loading of Au and CoOx respectively on electron-enriched {010} face and hole-enriched {110} face of BiVO4 nanocrystals [11]. However, there are few works reported on photodeposition of metal particles on oxide composite surfaces. Cu2O acts as a nice hole-scavenger co-catalyst for TiO2 [29-31]. In this letter, we report the structures and photocatalytic performances in water reduction of Pt/Cu2O-TiO2 composite photocatalysts prepared by photodeposition of Pt particles on Cu2O-TiO2 composite surfaces. The results reveal an interesting isoelectric point-controlled preferential adsorption and photodeposition of Pt species on Cu2O-TiO2 composite surfaces.
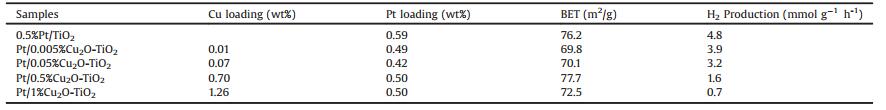
Experimental details are described in the Supporting Information. Pt with loadings of about 0.5% was photodeposited on TiO2 and Cu2O-TiO2 composites employing H2PtCl6 aqueous solutions as the Pt precursor. Under the photodeposition condition, the pH values of the solutions were measured to be 3.6. Table 1 summarizes compositions and BET specific surface areas of various photocatalysts. Both Pt loadings and BET specific surface areas are similar for all photocatalysts. Fig. 1 shows photocatalytic H2 productions as a function of reaction time of various photocatalysts in photocatalytic water reduction illuminated with simulated solar light. It can be seen that the photocatalytic H2 production increases linearly with the reaction time for various photocatalysts, indicating the stability of these photocatalysts. The calculated mass-specific photocatalytic H2 production rates of all photocatalysts are summarized in Table 1. 0.5%Pt/TiO2 exhibits a H2 production rate of 4.8 μmol g-1 h-1, but the H2 production rate of Pt-Cu2O-TiO2 composite photocatalysts keeps decreasing with the loading of Cu2O. These results suggest that the Pt and Cu2O species in Pt/Cu2O-TiO2 composite photocatalysts exert a negative effect, instead of a synergetic effect, on promoting the photocatalytic performance of TiO2.
|
|
Table 1 Compositions, BET specific surface areas and mass-specific photocatalytic H2 productions of various photocatalysts. |

|
Download:
|
| Fig. 1. Photocatalytic H2 production as a function of reaction time of various Pt/Cu2O-TiO2 under simulated solar light irradiation. | |
{kind=link}
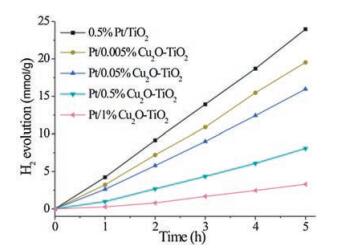
Fig. 2A shows XRD patterns of various photocatalysts, in which all observed diffraction patterns could be indexed to anatase TiO2 (JCPDS card: No. 21-1272). No peaks associated with Cu or Pt species could be identified. This may be due to the fine dispersions of copper and platinum species on TiO2 and/or the low loadings of copper and platinum in all photocatalysts. Surface structures of all photocatalysts were characterized with XPS. As shown in Fig. S1 (Supporting information), all Pt/Cu2O-TiO2 composite photocatalysts exhibit the same Ti 2p3/2 binding energy at 458.8 eV and Cu 2p3/2 binding energy at 932.4 eV that respectively correspond to TiO2 and Cu2O [30]. However, they exhibit Cu2O loadingdependent Pt 4f XPS features (Fig. 2B). A peak at around 75.6 eV corresponds to an energy loss peak of TiO2, and other peaks at 70.9/74.3, 72.8/76.1, and 74.2/77.5 eV can be assigned to the 4f7/2/4f5/2 components of metallic Pt, Pt2+, and Pt4+ species, respectively [32-34]. Fig. 2C shows the Pt speciation in various photocatalysts. 0.5% Pt/TiO2 exhibits dominant metallic Pt species with minor Pt2+ species. With the Cu2O loading of Pt/Cu2O-TiO2 increasing, the metallic Pt species decreases and could not be observed in Pt/1% Cu2O-TiO2; the Pt2+ species increases, reaches the maximum in Pt/0.5%Cu2O-TiO2 and then slightly decreases in Pt/1%Cu2O-TiO2; and the Pt4+ species emerges in Pt/1%Cu2O-TiO2. These XPS results demonstrate that the Pt species changes from the metallic Pt to the Pt cations in our Pt/Cu2O-TiO2 photocatalysts prepared by photodeposition as the Cu2O loading increases.

|
Download:
|
| Fig. 2. (A) XRD patterns, (B) Pt 4f XPS spectra with peak-fitting results, and (C) calculated Pt speciation of (a) 0.5%Pt/TiO2, (b) Pt/0.005%Cu2O-TiO2, (c) Pt/0.05%Cu2O-TiO2, (d) Pt/0.5%Cu2O-TiO2 and (e) Pt/1%Cu2O-TiO2 photocatalysts. The red line in Fig. 2A represents the standard XRD pattern of anatase TiO2 (JCPDS Card No. 21-1272). The scatter points and solid lines in Fig. 2B respectively represent the original XPS spectra and peak-fitted XPS spectra. | |
{kind=link}
Representative TEM and HRTEM images of Pt/TiO2 and Pt/Cu2OT-iO2 photocatalysts are shown in Fig. 3, Figs. S2 and S3 (Supporting information). The identified lattice spacings of 0.35, 0.21, 0.23, 0.26 and 0.22 nm arise from TiO2(101), Cu2O(200), Pt(111), PtO(101) and PtO2(011), respectively [29, 30, 35, 36]. Fig. S3 gives FT-transformed patterns of TRTEM images of individual particles to distinguish nanoparticles with similar observed lattice spacings. As reported in our previous paper [29], TiO2 exhibits a rod shape, and Cu2O forms a thin film on TiO2 in Cu2O-TiO2 composites up to 1%Cu2O-TiO2, forming TiO2 (core)/Cu2O (thin film shell) rod structures in which the Cu2O shell thickness increases with the Cu2O loading. Pt/TiO2 and Pt/Cu2O-TiO2 photocatalysts remain rod shapes, and Cu2O exists as thin layers on TiO2 in all Pt/Cu2O-TiO2 photocatalysts, but exposed TiO2 surfaces are always present. This is also confirmed by the DRIFTS results of CO adsorption on Pt/Cu2O-TiO2 photocatalysts (Fig. S4 in Supporting information) in which the vibrational band of CO adsorption at Ti(IV) sites is observed for Pt/Cu2O-TiO2 photocatalysts [37]. Thus the original TiO2 (core)/Cu2O (thin film shell) rod structures of Cu2O-TiO2 composites get destroyed after the photodeposition processes in H2PtCl6 aqueous solutions. Metallic Pt nanoparticles of 2–3 nm can be easily identified in Pt/TiO2, Pt/0.005%Cu2O-TiO2 and Pt/0.05%Cu2O-TiO2 but few PtO nanoparticles is found although the XPS results suggest the presence of minor PtO species. This could be attributed to the generally much higher dispersion of Pt oxides than Pt metal supported on oxide surfaces. In Pt/0.5%Cu2O-TiO2 and Pt/1%Cu2OT-iO2 photocatalysts with Pt oxides as the dominant Pt species, PtO and PtO2 nanoparticles of 3–4 nm are easily observed but Pt nanoparticles can only be occasionally found. It is found that the Pt species supported on TiO2 are always metallic Pt nanoparticles while those supported on Cu2O include metallic Pt nanoparticles in Pt/0.005%Cu2O-TiO2 and Pt/0.05%Cu2O-TiO2 but then are exclusively PtO and PtO2 nanoparticles in Pt/0.5%Cu2O-TiO2 and Pt/1% Cu2O-TiO2. Meanwhile, much more Pt species are photodeposited on the Cu2O surface of Cu2O-TiO2 composites than on the TiO2 surface. This agrees with the XPS results of dominant Pt oxides species since the photodeposited Pt species on TiO2 is mainly metallic Pt nanoparticles.

|
Download:
|
| Fig. 3. Representative TEM and HRTEM images of (A1 and A2) 0.5%Pt/TiO2, (B1, B2, B3) Pt/0.005%Cu2O-TiO2, (C1, C2, C3) Pt/0.05%Cu2O-TiO2, (D1, D2, D3) Pt/0.5%Cu2O-TiO2, and (E1, E2, E3) Pt/1%Cu2O-TiO2. Lattice spacings of 0.35, 0.21, 0.23, 0.26 and 0.22 nm correspond to TiO2(101), Cu2O(200), Pt(111), PtO(101) and PtO2(011), respectively. | |
{kind=link}
The above spectroscopic and microscopic characterization results demonstrate that the presence of Cu2O layers on TiO2 strongly affects the photodeposition processes of Pt. The photodeposited Pt species is preferentially formed on the Cu2O surface, and changes from metallic Pt nanoparticles to PtO and PtO2 nanoparticles as the Cu2O layers thicken. It is reasonable that the synthesized Pt/Cu2O-TiO2 photocatalysts with dominant PtO and PtO2 nanoparticles on Cu2O surface and few Pt nanoparticles on TiO2 surface are less active than the synthesized Pt/TiO2 catalyst with Pt nanoparticles in photocatalytic water reduction, as experimentally observed.
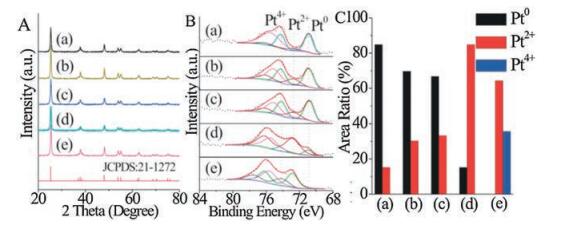
During the photodeposition process, the [PtCl4]2- precursor needs to adsorb on the oxide surface prior its photoreduction. Depending on its isoelectric point and the pH value of aqueous solution, an oxide surface in the aqueous solution is negatively, neutrally, or positively charged. We measured Zeta potentials of TiO2, Cu2O, and various Cu2O/TiO2 composites as a function of pH values (Fig. 4), from which the isoelectric point of TiO2, 0.005% Cu2O/TiO2, 0.05%Cu2O/TiO2, 0.5%Cu2O/TiO2, 1%Cu2O/TiO2 and commercial Cu2O is determined as 3.1, 4.8, 5.3, 5.8, 7.1 and 10.2, respectively. The pH value of employed H2[PtCl4] aqueous solution is measured to be 3.6, thus the TiO2 surface of Cu2O-TiO2 composites is locally negatively charged while the Cu2O surface is locally strongly positively charged. Based on these observations, we proposed a photodeposition mechanism of Pt on Cu2O-TiO2 composites (Fig. 5). Under the employed photodeposition conditions, the negatively-charged [PtCl4]2- precursor adsorb majorly on the strongly positively-charged Cu2O surface of Cu2O-TiO2 composites but minorly on the negatively-charged TiO2 surface, resulting in the preferential adsorption of [PtCl4]2-precursor on Cu2O surface of Cu2O-TiO2 composites. Illuminated with light, both Cu2O and anatase TiO2 are photoexcited to produce the electron-hole carriers, and the excited electrons in the conduction band of Cu2O tend to transfer to the conduction band of TiO2 at the Cu2O-TiO2 junction interface while the excited holes in the valence band of TiO2 tend to transfer to the valence band of Cu2O. This leads to electron-rich TiO2 and hole-rich Cu2O in Cu2OT-iO2 composites. Thus, the [PtCl4]2- species on electron-rich TiO2 surface can be facilely reduced to form metallic Pt nanoparticles while those on hole-rich Cu2O surface can not be adequately reduced and tend to form PtO nanoparticles and even PtO2 nanoparticles. The reduction extent of [PtCl4]2- species on Cu2O surface decreases as its thickness on TiO2 increases. Thus, oxide isoelectric point plays an important role in the photodeposition of Pt on Cu2O-TiO2 composite surfaces via preferential adsorption of [PtCl4]2- precursor. These reveal the important role of surface chemistry in photo-induced chemical processes on solid surfaces [38].

|
Download:
|
| Fig. 4. Zeta potentials of TiO2, Cu2O, and various Cu2O/TiO2 composites as a function of pH values of aqueous solution. | |
{kind=link}

|
Download:
|
| Fig. 5. Schematic illustration of the photodeposition process of H2PtCl6 on Cu2O/TiO2 composite. The yellow, red, blue spheres represent TiO2, Cu2O, Pt species, respectively. | |
{kind=link}
In summary, we have successfully investigated the photodeposition processes of Pt on Cu2O-TiO2 composite surfaces employing H2PtCl6 aqueous solution as the precursor and revealed a key role of oxide isoelectric point-controlled adsorption of [PtCl4]2- precursor in the photodeposition of Pt on Cu2O-TiO2 composites. Under the photodeposition conditions, the PtCl62-precursor adsorb dominantly on positively-charged and holeenriched Cu2O surface of Cu2O-TiO2 composites, but minorly on negatively-charged and electron-enriched TiO2 surface. This leads to the dominant formation of Pt oxide particles on Cu2O surface but few Pt metal particles on TiO2 surface. Consequently, the activity of resulting Pt/Cu2O-TiO2 composite photocatalysts in photocatalytic water reduction decreases as the Cu2O content increases. These results deepen the understanding of photodeposition processes on oxide composite surfaces.
AcknowledgmentsThis work was financially supported by the National Key R & D Program of Ministry of Science and Technology of China (No. 2017YFB0602205), the National Natural Science Foundation of China (Nos. 21525313, 91745202, 21703001) and the Changjiang Scholars Program of Ministry of Education of China.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.01.017.
| [1] |
A. Fujishima, K. Honnda, Nature 238 (1972) 37-38. DOI:10.1038/238037a0 |
| [2] |
T. Hisatomi, T. Minegishi, K. Domen, Bull. Chem. Soc. Jpn. 85 (2012) 647-655. DOI:10.1246/bcsj.20120058 |
| [3] |
T. Hisatomi, J. Kubota, K. Domen, Chem. Soc. Rev. 43 (2014) 7520-7535. DOI:10.1039/C3CS60378D |
| [4] |
F. Xiong, Z.M. Wang, Z.F. Wu, et al., Sci. China Chem. 62 (2019) 1-6. |
| [5] |
J. Sun, D.K. Zhong, D.R. Gamelin, Energy Environ. Sci. 3 (2010) 1252-1261. DOI:10.1039/c0ee00030b |
| [6] |
J.F. Zhu, M. Zäch, Curr. Opin. Colloid Interface Sci. 14 (2009) 260-269. DOI:10.1016/j.cocis.2009.05.003 |
| [7] |
N.L. Wu, M.S. Lee, Int. J. Hydrogen Energy 29 (2004) 1601-1605. DOI:10.1016/j.ijhydene.2004.02.013 |
| [8] |
K. Lalitha, J.K. Reddy, M.V.P. Sharma, V.D. Kumari, M. Subrahmanyam, Int. J. Hydrogen Energy 35 (2010) 3991-4001. DOI:10.1016/j.ijhydene.2010.01.106 |
| [9] |
T.T. Le, M.S. Akhtar, D.M. Park, J.C. Lee, O.B. Yang, Appl. Catal. B-Environ. 111 (2012) 397-401. |
| [10] |
Y. Lin, Z.Y. Jiang, C.Y. Zhu, et al., Int. J. Hydrogen Energy 38 (2013) 5209-5214. DOI:10.1016/j.ijhydene.2013.02.079 |
| [11] |
Y. Qi, Y. Zhao, Y.Y. Gao, et al., Joule 2 (2018) 1-10. DOI:10.1016/j.joule.2017.10.014 |
| [12] |
R.T. Chen, S. Pang, H.Y. An, et al., Nat. Energy 3 (2018) 655-663. DOI:10.1038/s41560-018-0194-0 |
| [13] |
J.K. Zhang, Z.B. Yu, Z. Gao, et al., Angew. Chem. Int. Ed. 56 (2017) 816-820. DOI:10.1002/anie.201611137 |
| [14] |
Q. Zhang, Z. Li, S.Y. Wang, et al., ACS Catal. 6 (2016) 2182-2191. DOI:10.1021/acscatal.5b02503 |
| [15] |
J.H. Yang, D.E. Wang, H.X. Han, C. Li, Acc. Chem. Res. 46 (2013) 1900-1909. DOI:10.1021/ar300227e |
| [16] |
J.R. Ran, J. Zhang, J.G. Yu, M. Jaroniec, S.Z. Qiao, Chem. Soc. Rev. 43 (2014) 7787-7812. DOI:10.1039/C3CS60425J |
| [17] |
R.G. Li, F.X. Zhang, D.E. Wang, et al., Nat. Commun. 4 (2013) 1432. DOI:10.1038/ncomms2401 |
| [18] |
S. Mubeen, J. Lee, N. Singh, et al., Nat. Nanotechnol. 8 (2013) 247. DOI:10.1038/nnano.2013.18 |
| [19] |
D.A. Wang, T. Hisatomi, T. Takata, et al., Angew. Chem. Int. Ed. 52 (2013) 11252-11256. DOI:10.1002/anie.v52.43 |
| [20] |
B.J. Ma, F.Y. Wen, H.F. Jiang, et al., Catal. Lett. 134 (2010) 78-86. DOI:10.1007/s10562-009-0220-8 |
| [21] |
A.A. Ismail, D.W. Bahnemann, S.A. Al-Sayari, Appl. Catal. A 431 (2012) 62-68. |
| [22] |
É. Karácsonyi, L. Baia, A. Dombi, et al., Catal. Today 208 (2013) 19-27. DOI:10.1016/j.cattod.2012.09.038 |
| [23] |
M. Maicu, M. Hidalgo, G. Colón, J.A. Navío, J. Photochem. Photobiol. 217 (2011) 275-283. DOI:10.1016/j.jphotochem.2010.10.020 |
| [24] |
T. Sano, N. Negishi, K. Uchino, et al., J. Photochem. Photobiol. A 160 (2003) 93-98. DOI:10.1016/S1010-6030(03)00226-0 |
| [25] |
G.M. Ma, J.Y. Liu, T. Hisatomi, et al., Chem. Commun. 51 (2015) 4302-4305. DOI:10.1039/C4CC10297E |
| [26] |
Z. Jiang, Z.Y. Zhang, W.F. Shangguan, et al., Catal. Sci. Technol. 6 (2016) 81-88. DOI:10.1039/C5CY01364J |
| [27] |
B. Kraeutler, A.J. Bard, J. Am. Chem. Soc. 100 (1978) 4317-4318. DOI:10.1021/ja00481a059 |
| [28] |
R.W. Liang, F.F. Jing, L.J. Shen, N. Qin, L. Wu, Nano Res. 8 (2015) 37-3249. |
| [29] |
Y.X. Liu, B.S. Zhang, L.F. Luo, et al., Angew. Chem. Int. Ed. 127 (2015) 15475-15480. DOI:10.1002/ange.201509115 |
| [30] |
Y.X. Liu, Z.L. Wang, W.X. Huang, Appl. Surf. Sci. 389 (2016) 760-767. DOI:10.1016/j.apsusc.2016.07.173 |
| [31] |
Z.L. Wang, Y.X. Liu, D.J. Martin, et al., Phys. Chem. Chem. Phys. 15 (2013) 14956-14960. DOI:10.1039/c3cp52496e |
| [32] |
T.B. Zhou, H. Wang, S. Ji, V. Linkov, R.F. Wang, J. Power Sources 248 (2014) 427-433. DOI:10.1016/j.jpowsour.2013.09.108 |
| [33] |
D.P. He, Y.L. Jiang, H.F. Lv, M. Pan, S.C. Mu, Appl. Catal. B 132 (2013) 379-388. |
| [34] |
S. Liang, Y.M. Zhou, W.T. Wu, et al., J. Photochem. Photobiol. A 346 (2017) 168-176. DOI:10.1016/j.jphotochem.2017.06.005 |
| [35] |
L.H. Yu, Y. Shao, D.Z. Li, Appl. Catal. B 204 (2017) 216-223. DOI:10.1016/j.apcatb.2016.11.039 |
| [36] |
X.Y. Yang, Y. Li, P. Zhang, et al., ACS Appl. Mat. Interfaces 10 (2018) 23154-23162. DOI:10.1021/acsami.8b06815 |
| [37] |
D. Li, S.L. Chen, R. You, et al., J. Catal. 368 (2018) 163-171. DOI:10.1016/j.jcat.2018.09.032 |
| [38] |
W.X. Huang, Z.L. Wu, J.W. Tang, W. Wei, X.F. Guo, Chin. Chem. Lett. 29 (2018) 725-726. DOI:10.1016/j.cclet.2018.05.021 |