 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of the Discovery and Development of Novel Pesticide, Shenyang Sinochem Agrochemicals Research and Development Co., Ltd., Shenyang 110021, China;
c School of Life Science and Biotechnology, Dalian University of Technology, Dalian 116024, China
Chitin is the second most abundant biopolymer next to cellulose in nature, and is the major component of the extracellular matrix of some agricultural pests such as fungi [1], nematodes and insects [2]. Notably, chitin is completely absent from vertebrates and higher plants, and the key enzymes for chitin biosynthesis and biodegradation represent potential targets for pesticide development. For example, insect chitinolytic β-N-acetyl-D-hexosaminidase (Hex), which is responsible for hydrolyzing chitooligosaccharides to N-acetyl-D-glucosamine during chitin degradation, is considered to be an attractive target for pesticide development. Hex has been proved to be vital for insect survival in different insect species, such as Tribolium castaneum [3], Ostrinia furnacalis [4], Locusta migratoria [5], Nilaparvata lugens [6] and Mamestra brassicae [7]. Moreover, the crystal structure of OfHex1 from Ostrinia furnacalis has been resolved and has provided a solid basis for the design of specific inhibitors [8].
Several kinds of OfHex1 inhibitors have been reported, including TMG-chitotriomycin [9-14], PUGNAc [15, 16], NAG-thiazoline and itsderivativeNMAGT[17, 18], Q2[19], phlegmacinB1[20], berberine [21] and thiazolylhydrazone derivatives [22]. Among these compounds, Q2, an unsymmetrical dyad of thiadiazole and 1, 8- naphthalimide, is of great interest because it is the first noncarbohydrate inhibitor of OfHex1 [19]. As part of our efforts toward the discovery and biological evaluation of new OfHex1 inhibitor, we report here the synthesis and structure-activity relationships of a series of Q2 derivatives formed by replacing the thiadiazole and naphthalimide groups [23-25], and altering the linker length. Moreover, to improve the pharmacological profiles and solubilities of naphthalimide derivatives while maintaining their inhibitory activity, we replaced the naphthalimide moiety with quinoline carboxamide group via an intramolecular N-H hydrogen bond to mimic the naphthalimide configuration. We studied the inhibition mechanisms of these compounds using structure-based molecular docking.
The preparation of substituted naphthalimide derivatives 3a-m is summarized in Scheme 1. The dimethylamine-substituted intermediate 1b can be obtained from the corresponding commercially available 4-bromo-1, 8-naphthalic anhydride 1a. Intermediates 2 were synthesized by reacting a suspension of intermediates 1 in ethanol with excess Boc-protected amines, followed by deprotection in acidic condition. Compounds 3 were synthesized from intermediates 2 with a halogenated compound or aryl acyl chloride under base conditions [26].

|
Download:
|
| Scheme 1. Synthesis of compounds 3a-m. Reagents and conditions: (a) 40% Dimethylamine aqueous solution, CuSO4·5H2O, DMF, reflux, 8 h; (b) NH2(CH2)nNHBoc, EtOH, reflux, 3 h; (c) CF3COOH, CH2Cl2, r.t., 4 h; (d) R1Cl, K2CO3, CH3CN, reflux, 3 h; or R1Cl, Et3N, CH2Cl2, r.t., 5 h. | |
The quinoline derivatives were prepared according the method described in Scheme 2. The quinoline acyl chloride 4, which was prepared from quinoline carboxylic acid, was reacted with the Bocprotected amine and then deprotected under acidic condition to obtain the intermediate 5. Compounds 6 were synthesized from intermediate 5 with the halogenated compounds or aryl acyl chloride under base conditions. Details for the synthetic procedures, physical characteristics and the results of 1H NMR, 13C NMR and MS for all the synthesized compounds are listed in the Supporting information.

|
Download:
|
| Scheme 2. Synthesis of compounds 6a-j. Reagents and conditions: (a) SOCl2, PhCH3, reflux, 2 h; (b) tert-butyl(2-aminoethyl)carbamate, Et3N, CH2Cl2, r.t., 5 h; (c) CF3COOH, CH2Cl2, r.t., 4 h; (d) R3Cl, K2CO3, CH3CN, reflux, 3 h; or R3Cl, Et3N, CH2Cl2, r.t., 5 h. | |
The inhibitory activities of naphthalimide derivatives 3a-m toward OfHex1 are outlined in Table 1. For substitution of NH (R1), a comparison of 3a with 3b showed that small rings, such as thiazole, gave better inhibitory activity than the pyridine ring. The carbonyl group was introduced to increase the hydrogen bond between the compound and the enzyme, which could improve binding affinity. However, a comparison of 3a with 3c–i showed that replacement of methylene with a carbonyl or sulfone group did not remarkably improve the inhibitory activity.
|
|
Table 1 Inhibitory activities of compounds 3a-m against OfHex1. |
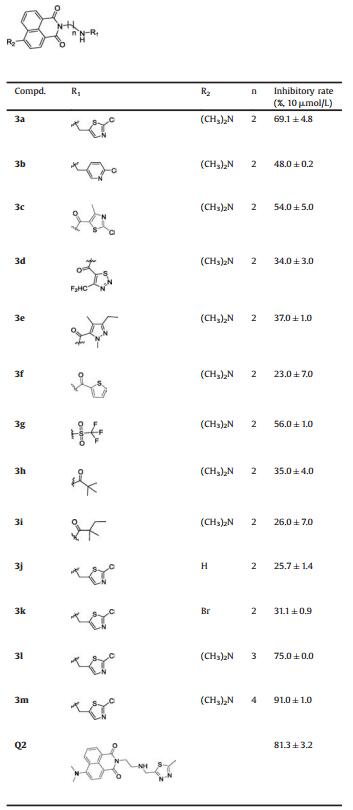
Compound 3a had an acceptable level of inhibitory activity, and the raw material of 3a, 2-chloro-5-(chloromethyl)thiazole, is cheap and easy to get. Thus, we used (2-chlorothiazol-5-yl)methyl as R1 and focused our attention on the effects of the substitution of the naphthalimide and elongation of the alkyl chain (n). Replacement of the dimethylamino group with bromine or hydrogen at the 5-position (R2; compound 3j and 3k) decreased the activity against OfHex1. A comparison of the inhibitory activities of compounds 3a, 3l and 3m showed a large enhancement with elongation of the alkyl chain (n). Compound 3m stood out with comparatively higher activity with a Ki value of 0.34 μmol/L (Fig. 1A), and exhibited about 4-fold increase in inhibitory activity when compared with compound Q2 (Ki = 1.4 μmol/L).

|
Download:
|
| Fig. 1. Inhibitory kinetics of compounds 3m (A), 6a (B) against OfHex1. | |
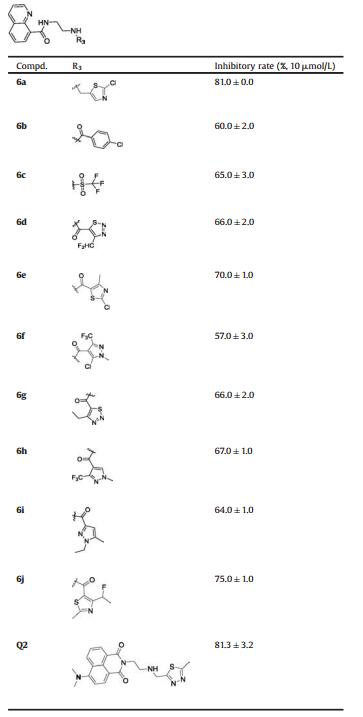
To improve the pharmacological profiles of naphthalimide derivatives but maintain the inhibitory activity, we replaced the naphthalimide moiety with quinoline carboxamide group, which contains an intramolecular N-H hydrogen bond and could mimic the naphthalimide while improving the solubility. The quinoline analogues showed obvious better inhibitory activities against OfHex1, regardless of their content of a six-membered ring or fivemembered ring (Table 2). Compounds containing a thiazole ring, including 6a, 6e and 6j, exhibited more potent inhibitory activity (>70%) than compounds containing other heterocycle ring. Compound 6a showed the best inhibitory activity with a Ki value of 2.3 μmol/L against OfHex1 (Fig. 1B).
|
|
Table 2 Inhibitory activities of compounds 6a-j against OfHex1. |
{kind=link}
{kind=link}
{kind=link}
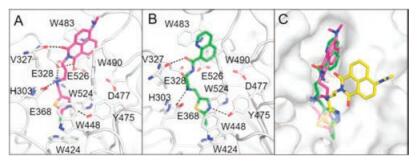
To elucidate the inhibition mechanism of 3m, molecular docking was performed using OfHex1 as template. We found that 3m occupied the entire substrate binding pocket of OfHex1 and interacted via hydrogen bonds (Fig. 2A). The molecular docking study revealed good binding of the linker and the thiazole group of 3m at the subsite -1. The linker of 3m was bent into a curved conformation and the secondary nitrogen atom formed a hydrogen bond with the catalytic residue Glu368 and the residue Glu328. The N3 atom formed a hydrogen bond with the phenolic hydroxyl group of Tyr475. Interestingly, the length of the linker region had a strong effect on the inhibition mechanism. Elongation of the alkyl chain resulted in tight binding of the 4-dimethylaminonaphthalimide group of 3m. A hydrophobic patch comprising Trp483, Trp490 and carbonyl groups from the 4-dimethylaminonaphthalimide formed hydrogen bonds with Val327, Glu328 and Glu526.

|
Download:
|
| Fig. 2. Inhibition mechanisms of compounds 3m and 6a against OfHex1. (A) Binding mode of 3m in the active pocket of OfHex1. Compound 3m was shown in magenta. The hydrogen bonds were shown in black dashes. (B) Binding mode of 6a in the active pocket of OfHex1. Compound 6a was shown in green. The hydrogen bonds are shown as dashed black lines. (C) Superimposition of compound 3m, 6a and Q2 in the active pocket of OfHex1. 3m, 6a and Q2 are shown in magenta, green and yellow respectively. | |
{kind=link}
The binding mode of compound 6a in OfHex1 was studied by molecular docking. As shown in Fig. 2B, binding of compound 6a occurred in the entire active pocket of OfHex1 in a similar manner to compound 3m. The mechanisms of interaction of the linkers and thiazole group with OfHex1 were similar to those of 3m, and the quinoline group bound with a hydrophobic patch comprising Trp483 and Trp490 outside of subsite -1.
Although compounds 3m (Ki = 0.34 μmol/L) and 6a (Ki = 2.3 μmol/L) showed different inhibitory activity as Q2 did (Ki = 1.4 μmol/L) against OfHex1, the predicted binding modes of 3m and 6a were similar to that of Q2 in the crystal structure of OfHex1 in a complex with Q2 [19] (Fig. 2C). First, binding of the thiazole group of 3m, 6a and the thiadiazole group of Q2 occurred in subsite -1 of the active pocket in the same fashion. These groups were sandwiched by Trp524 and Trp448 and their N3 atoms formed hydrogen bonds with the phenolic hydroxyl group of Tyr475. Second, the linkers of 3m, 6a and Q2 were bent into a curved conformation and the secondary nitrogen atoms formed hydrogen bonds with the catalytic residue Glu368. These results suggest that the compounds discovered in this study inhibit OfHex1 by a similar mechanism as Q2.
In summary, we designed, prepared and evaluated a series of substituted naphthalimide and quinoline derivatives as potential inhibitors of OfHex1. Compound 3m was the most potent inhibitor with a Ki value of 0.34 μmol/L. Quinoline analogs with an intramolecular N-H hydrogen bond mimic the naphthalimide configuration to maintain the inhibitory activity potency.
AcknowledgmentsThe authors acknowledge the National Key Research and Development Program of China (Nos. 2017YFD0200500, 2017YFD0201400, 2018YFD0200100) and the National Natural Science Foundation of China(No.31871959)for the financial support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.01.023.
| [1] |
V.G.H. Eijsink, G. Vaaje-Kolstad, K.M. Varum, S.J. Horn, Trends Biotechnol. 26 (2008) 228-235. DOI:10.1016/j.tibtech.2008.02.004 |
| [2] |
H. Merzendorfer, L. Zimoch, J. Exp. Biol. 206 (2003) 4393-4412. DOI:10.1242/jeb.00709 |
| [3] |
D.G. Hogenkamp, Y. Arakane, K.J. Kramer, S. Muthukrishnan, R.W. Beeman, Insect Biochem. Mol. Biol. 38 (2008) 478-489. DOI:10.1016/j.ibmb.2007.08.002 |
| [4] |
T. Liu, H. Zhang, F. Liu, et al., J. Biol. Chem. 286 (2011) 4049-4058. DOI:10.1074/jbc.M110.184796 |
| [5] |
S. Rong, D.Q. Li, X.Y. Zhang, et al., Insect Sci. 20 (2013) 109-119. DOI:10.1111/j.1744-7917.2012.01573.x |
| [6] |
Y. Xi, P.L. Pan, C.X. Zhang, Insect Mol. Biol. 24 (2015) 601-610. DOI:10.1111/imb.2015.24.issue-6 |
| [7] |
H. Zhang, K.J. Zhao, D.J. Fan, Asia-Pac. Entomol. 19 (2016) 721-728. DOI:10.1016/j.aspen.2016.06.012 |
| [8] |
Q. Yang, T. Liu, F. Liu, M. Qu, X. Qian, FEBS J. 275 (2008) 5690-5702. DOI:10.1111/j.1742-4658.2008.06695.x |
| [9] |
H. Usuki, T. Nitoda, M. Ichikawa, et al., J. Am. Chem. Soc. 130 (2008) 4146-4152. DOI:10.1021/ja077641f |
| [10] |
Y. Yang, Y. Li, B. Yu. J. Am. Chem. Soc. 131 (2009) 12076-12077. DOI:10.1021/ja9055245 |
| [11] |
Y. Yang, T. Liu, Y. Yang, et al., Chem. Bio. Chem. 12 (2011) 457-467. DOI:10.1002/cbic.v12.3 |
| [12] |
H. Usuki, Y. Yamamoto, Y. Kumagai, et al., Org. Biomol. Chem. 9 (2011) 2943-2951. DOI:10.1039/c0ob01090a |
| [13] |
S. Halila, E. Samain, C.E. Vorgias, S. Armand, Carbohydr. Res. 368 (2013) 52-56. DOI:10.1016/j.carres.2012.12.007 |
| [14] |
Y. Isoda, N. Sasaki, K. Kitamura, et al., Beilstein J. Org. Chem. 13 (2017) 919-924. DOI:10.3762/bjoc.13.93 |
| [15] |
M. Horsch, L. Hoesch, A. Vasella, D.M. Rast, FEBS J. 197 (1991) 815-818. |
| [16] |
T. Liu, H. Zhang, F. Liu, et al., Biochem. J. 438 (2011) 467-474. DOI:10.1042/BJ20110390 |
| [17] |
S. Knapp, D. Vocadlo, Z. Gao, et al., J. Am. Chem. Soc. 118 (1996) 419-424. |
| [18] |
T. Liu, M. Xia, H. Zhang, et al., FEBS Lett. 589 (2015) 110-116. DOI:10.1016/j.febslet.2014.11.032 |
| [19] |
T. Liu, P. Guo, Y. Zhou, et al., Sci. Rep. 4 (2014) 6188. |
| [20] |
L. Chen, T. Liu, Y. Duan, X. Lu, Q. Yang, J. Agric. Food Chem. 65 (2017) 3851-3857. DOI:10.1021/acs.jafc.7b01710 |
| [21] |
Y. Duan, T. Liu, Y. Zhou, T. Dou, Q. Yang, J. Biol. Chem. 293 (2018) 15429-15438. DOI:10.1074/jbc.RA118.004351 |
| [22] |
H. Yang, T. Liu, H. Qi, et al., Bioorg. Med. Chem. 26 (2018) 5420-5426. DOI:10.1016/j.bmc.2018.09.014 |
| [23] |
Z. Chen, Y. Xu, X. Qian, Chin. Chem. Lett. 29 (2018) 1741-1756. DOI:10.1016/j.cclet.2018.09.020 |
| [24] |
S. Zhou, Y. Xie, X. Meng, et al., Chin. Chem. Lett. 29 (2018) 1254-1256. DOI:10.1016/j.cclet.2017.10.022 |
| [25] |
L. Zhou, Z. Jin, X. Fan, et al., Chin. Chem. Lett. 29 (2018) 1500-1502. DOI:10.1016/j.cclet.2018.07.018 |
| [26] |
P. Guo, Q. Chen, L. Xu, Q. Yang, X. Qian, ACS Med. Chem. Lett. 4 (2013) 527-531. DOI:10.1021/ml300475m |