 2019, Vol. 30
2019, Vol. 30 


b Department of Chemistry, Innovative Drug Research Center, Shanghai University, Shanghai 200444, China
Organofluorine compounds are widely employed in pharmaceuticals, agrochemicals and materials due to their extraordinary physical and chemical properties [1]. Specifically, introducing the trifluoromethyl (CF3) moiety into biologically active molecules has shown to dramatically improve metabolic stability, lipophilicity and membrane permeability [1, 2]. As an effective strategy in drug modification, the development of an economical and efficient protocol to install the trifluoromethyl group into organic molecules is highly desirable. In the past decade, a number of transitionmetal-catalyzed trifluoromethyl-ation reactions have been reported [3]. Notably, transition-metal-catalyzed C–H activation to introduce the CF3 moiety would be the most efficient method [4]. Sanford, Yu, Shi et al. have reported the palladium-catalyzed direct C–H trifluoromethylation of arenes by Pd(Ⅱ)/Pd(Ⅳ) redox catalysis [5]. In addition to this, copper-catalyzed/mediated [6] and silver-mediated [7] C–H trifluoromethylation of arenes have also been reported.
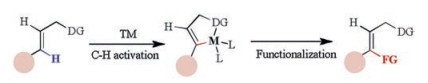
Recently, trifluoromethylation of alkenes has drawn more and more attentions. Many synthetic methods have been developed including cross-coupling reactions of trifluoromethylation reagent with vinyl halides [8], vinyl sulfonates [9] and vinyl borons [10] or decarboxylative trifluoromethylation [11]. However, direct C–H trifluoromethylations of alkenes are rarely reported. Generally, during metal-catalyzed directed C–H activation of alkenes, five membered cyclometalation intermediates are formed, resulting in C–H functionalization selectively at the β-position (Scheme 1) [12]. Loh [13] and Besset [14] have reported the Cu-catalyzed or mediated β-C–H trifluoromethylation of alkenes, independently. Recently, our group developed a Pd-catalyzed directed α-selective olefinic C–H activation to construct 4-imino-lactam derivatives (Scheme 2a) [15]. In addition, Bi reported a copper-catalyzed α-selective trifluoromethylation of α, β-unsaturated carbonyl compounds with Togni's reagent (Scheme 2b) [16]. To the best of our knowledge, the transition-metal catalyzed or mediated C–H trifluoromethylation of alkenes involved the electrophilic trifluoromethylation reagents, such as Togni [17] or Umemoto reagents [18]. Promoted by our interest in copper-mediated ortho C–H trifluoromethylation [19] and other transformations [20], we questioned whether a catalytic C–H trifluoromethylation of alkenes with readily available nucleophilic trifluoromethylation reagents such as Ruppert-Prakash regent (TMSCF3, TMS = trimethylsilyl) could be designed. If successful, such a platform might offer a new approach for the preparation of trifluoromethyl-substituted olefin. Herein, we reported a Cu (Ⅱ)-catalyzed direct α-selective C–H trifluoromethylation of β-substituted acrylamides with TMSCF3 (Scheme 2c).

|
Download:
|
| Scheme 1. Metal-catalyzed directed C–H activation of alkene. | |

|
Download:
|
| Scheme 2. Catalytic-olefinic C–H functionalization. | |
We began our investigation with N-phenyl or alkyl substituted cinnamamide, and found N-isopropyl substituted cinnamamide gave the best result (Table S1 in Supporting information). Treatment cinnamamide 1a with 5 equiv. of TMSCF3 in the presence of 1 equiv. of Cu(OAc)2, 1.5 equiv. of Ag2CO3 and 4 equiv. of KF in DMSO at 80 ℃ afforded the α-selective C–H trifluoromethylation compounds in 29% yield. Ag2CO3 played an important role in the reaction system, and the yield reduced significantly without Ag2CO3 (Table S5 in Supporting information). Exceptionally, the cinnamic acid did not show any reactivity. Increasing the temperature to 120 ℃ for 6 h, the yield was improved to 41%.
We also examined the influence of base, and found LiF and KH2PO4 shown the best result (Table 1, entries 1–6). Pleasingly, lowering the amount of Cu(OAc)2 to 20 mol% has little effect on the yield (Table 1, entries 7 and 8), indicating that the reaction could proceed catalytically.Increasing the loading of Ag2CO3 to 2 equiv. could improve the yields to 61% within 30 min (Table 1, entry 9). Ligands have been proved to promote the copper-catalyzed C–H functionalization [21], thus various ligands were added to improve the yield (Table S7 in Supporting information). To our delight, further addition of 20 mol% pyridine ligand could improve the yield to 73% (Table 1, entries 10 and 11).
|
|
Table 1 Reaction optimization.a |
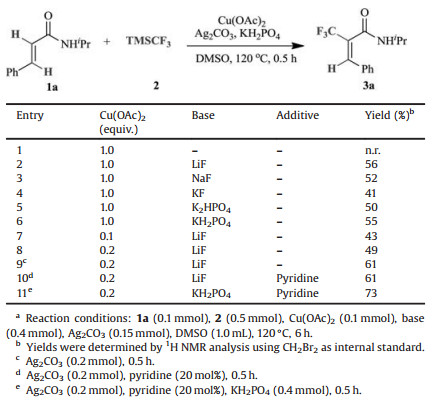
{kind=link}
{kind=link}
With the optimized conditions in hand, we turned our attention to explore the generality of the Cu-catalyzed α-selective C–H trifluoromethylation of acrylamides with TMSCF3. As shown in Scheme 3, a wide range of cinnamamide substrates with both electron-rich (1a-1f) and electron-deficient substituents (1i, 1j, 1p, 1q) were tolerated, affording the trifluoromethylation products in moderate to good yields. The trifluoromethoxy (1g, 1h) and trifluoromethyled substrates (1i, 1j) gave the desired products in good yields under the standard conditions, thus giving access to multiple fluorine containing compounds. Notably, the halogen containing substrates (1k-1o) were well tolerant, leaving an additional functional group for further transformation. Additionally, the cyano and nitro substituted cinnamamide (1p, 1q) could be employed in the reaction, affording the products in 57% and 60% yields respectively. Encouraged by previous results, we wondered whether our protocol could be extended to heterocycle-containing compounds. By far now, the copper-catalyzed olefinic trifluoromethylation of heterocycle-based substrates remains a challenge [22]. We were pleased to find that the pyridyl (1r-1t), thienyl (1u) and thiazolyl (1v) substituted acrylamides were all compatible in the reaction, thus allowing for the construction of the corresponding trifluoromethylated products in moderate yields.

|
Download:
|
| Scheme 3. Scope of substrates. Reaction conditions: 1a-1v (0.2 mmol), 2 (1.0 mmol), Cu(OAc)2 (20 mol%), pyridine (20 mol%), Ag2CO3 (0.4 mmol), KH2PO4 (0.8 mmol), DMSO (2 mL), 120 ℃, 0.5 h. Yield means isolated yield. | |
{kind=link}
To further demonstrate the utility of our methodology, we conducted the reaction on a gram scale and provided 3a in 56% yield (Scheme 4).

|
Download:
|
| Scheme 4. Scale up reaction. | |
{kind=link}
Control experiments were carried out to explore the reaction mechanism. As shown in Scheme 5, reaction a, copper is essential in the reaction and no reaction occurred without copper catalyst. The yields dropped significantly to 10% in the absence of Ag2CO3. Additionally, we conducted radical trapping experiments under the standard conditions as shown in Scheme 5, reaction b. The commonly used radical quencher 2, 2, 6, 6-tetramethylpiperidine-N-oxyl (TEMPO) and 2, 6-di-tert-butyl-4-methylphenol (BHT) inhibited the reaction completely, which indicated a radical pathway or a single electron transfer (SET) mechanism may be involved in the reaction. However, the radical-trapped adduct (TEMPO-CF3) was not detected by GC–MS and NMR analysis of the crude reaction mixture [23]. Based on the control experiments and literature precedents, we proposed a plausible radical mechanism [16, 24]. As shown in Scheme 5, reaction c, CF3 radical is generated from TMSCF3 through an AgCF3 species [7a, 7b, 20]. The CF3 radical then attacks the α-position of 1 that has a slightly higher electron density, thus forming the radical intermediate A. Subsequently, the carbocation intermediate B is formed by Cu(Ⅱ) through the SET process. Finally, the more stable (E)-configuration product 3 is obtained after deprotonation.

|
Download:
|
| Scheme 5. (a) Control experiments, (b) radical trapping experiments, (c) proposed mechanism. | |
{kind=link}
In conclusion, we have developed Cu(OAc)2 catalyzed α-selective C—H trifluoromethylation of acrylamide with TMSCF3 as nucleophilic CF3 source. This protocol is characterized by its exceedingly fast reaction rate with excellent regioselectivity and good functional group compatibility. Further investigations into related processes and understanding of the mechanistic intricacies are currently ongoing in our laboratories.
AcknowledgmentsWe gratefully acknowledge the National Natural Science Foundation of China (Nos. 21472211, 21502212, 21772211), Youth Innovation Promotion Association CAS (Nos. 2014229 and 2018293), Institutes for Drug Discovery and Development, Chinese Academy of Sciences (No. CASIMM 0120163006), Science and Technology Commission of Shanghai Municipality (No. 17JC1405000) for financial support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.02.011.
| [1] |
(a) P.Jeschke, ChemBioChem.5(2004)570-589; (b) K.Muller, C.Faeh, F.Diederich, Science 317(2007)1881-1886; (c) S.Purser, P.R.Moore, S.Swallow, V.Gouverneur, Chem.Soc.Rev.37(2008)320-330. |
| [2] |
(a) R.Filler, Y.Kobayashi, L.M.Yagupolskii, Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications, Elsevier, New York, 1993; (b) W.K.Hagmann, J.Med.Chem.51(2008)4359-4369. |
| [3] |
(a) T.Furuya, A.S.Kamlet, T.Ritter, Nature 473(2011)470-477; (b) G.Landelle, A.Panossian, S.Pazenok, J.P.Vors, F.R.Leroux, Beilstein J.Org.Chem.9(2013)2476-2536; (c) L.Chu, F.L.Qing, Acc.Chem.Res.47(2014)1513-1522; (d) G.B.Li, C.Zhang, C.Song, Y.D.Ma, Beilstein J.Org.Chem.14(2018)155-181; (e) Y.Guo, M.W.Huang, X.L.Fu, et al., Chin.Chem.Lett.28(2017)719-728; (f) T.Besset, T.Poisson, X.Pannecoucke, Chem.-Eur.J.20(2014)16830-16845; (g) E.Merino, C.Nevado, Chem.Soc.Rev.43(2014)6598-6608; (h) H.Egami, M.Sodeoka, Angew.Chem.Int.Ed.53(2014)8294-8308. |
| [4] |
(a) H.Liu, Z.Gu, X.Jiang, Adv.Synth.Catal.355(2013)617-626; (b) C.Alonso, E.Martínez de Marigorta, G.Rubiales, F.Palacios, Chem.Rev.115(2015)1847-1935; (c) X.Wang, Y.Ye, G.Ji, et al., Org.Lett.15(2013)3730-3733; (d) N.O.Ilchenko, P.G.Janson, K.J.Szabó, Chem.Commun.49(2013)6614-6616. |
| [5] |
(a) Y.Ye, N.D.Ball, J.W.Kampf, M.S.Sanford, J.Am.Chem.Soc.132(2010)14682-14687; (b) X.Wang, L.Truesdale, J.Q.Yu, J.Am.Chem.Soc.132(2010)3648-3649; (c) X.G.Zhang, H.X.Dai, M.Wasa, J.Q.Yu, J.Am.Chem.Soc.134(2012)11948-11951; (d) M.Miura, C.G.Feng, S.Ma, J.Q.Yu, Org.Lett.15(2013)5258-5261; (e) L.S.Zhang, K.Chen, G.Chen, et al., Org.Lett.15(2013)10-13. |
| [6] |
(a) L.Chu, F.L.Qing, J.Am.Chem.Soc.134(2012)1298-1304; (b) S.J.Cai, C.Chen, Z.L.Sun, C.J.Xi, Chem.Commun.49(2013)4552-4554. |
| [7] |
(a) Y.D.Ye, S.H.Lee, M.S.Sanford, Org.Lett.13(2011)5464-5467; (b) A.Hafner, S.Brase, Angew.Chem., Int.Ed.51(2012)3713-3715; (c) S.Seo, J.B.Taylor, M.F.Greaney, Chem.Commun.49(2013)6385-6387; (d) G.F.Shi, C.D.Shao, S.L.Pan, J.X.Yu, Y.H.Zhang, Org.Lett.17(2015)38-41 and references therein.. |
| [8] |
(a) Y.Kobayashi, K.Yamamoto, I.Kumadaki, Tetrahedron Lett.20(1979)4071-4072; (b) T.Kitazume, N.Ishikawa, J.Am.Chem.Soc.107(1985)5186-5191; (c) J.X.Duan, D.B.Su, Q.Y.Chen, J.Fluorine Chem.61(1993)279-284; (d) A.Hafner, S.Bräse, Adv.Synth.Catal.353(2011)3044-3048. |
| [9] |
E.J. Cho, S.L. Buchwald, Org.Lett. 13 (2011) 6552-6555. DOI:10.1021/ol202885w |
| [10] |
(a) L.Chu, F.L.Qing, Org.Lett.12(2010)5060-5063; (b) J.Xu, D.F.Luo, B.Xiao, et al., Chem.Commun.47(2011)4300-4302;( ) T.Liu, Q.Shen, Org.Lett.13(2011)2342-2345; (d) A.T.Parsons, T.D.Senecal, S.L.Buchwald, Angew.Chem.Int.Ed.51(2012)2947-2950; (e) Y.Li, L.Wu, H.Neumann, M.Beller, Chem.Commun.49(2013)2628-2630; (f) M.Presset, D.Oehlrich, F.Rombouts, G.A.Molander, J.Org.Chem.78(2013)12837-12843; (g) S.Arimori, N.Shibata, Org.Lett.17(2015)1632-1635. |
| [11] |
(a) Z.He, T.Luo, M.Hu, Y.Cao, J.B.Hu, Angew.Chem.Int.Ed.51(2012)3944-3947; (b) Z.J.Li, Z.L.Cui, Z.Q.Liu, Org.Lett.15(2013)406-409; (c) T.Patra, A.Deb, S.Manna, U.Sharma, D.Maiti, Eur.J.Org.Chem.(2013)5247-5250. |
| [12] |
K. Wang, F. Hu, Y. Zhang, J. Wang, Sci.China Chem. 58 (2015) 1252-1265. DOI:10.1007/s11426-015-5362-5 |
| [13] |
(a) C.Feng, T.P.Loh, Chem.Sci.3(2012)3458-3462; (b) C.Feng, T.P.Loh, Angew.Chem.Int.Ed.52(2013)12414-12417. |
| [14] |
T. Besset, D. Cahard, X. Pannecoucke, J.Org.Chem. 79 (2014) 413-418. DOI:10.1021/jo402385g |
| [15] |
W.J. Kong, Y.J. Liu, H. Xu, et al., J.Am.Chem.Soc. 138 (2016) 2146-2149. DOI:10.1021/jacs.5b13353 |
| [16] |
Z. Fang, Y. Ning, P. Mi, P. Liao, X. Bi, Org.Lett. 16 (2014) 1522-1525. DOI:10.1021/ol5004498 |
| [17] |
(a) S.Barata-Vallejo, B.Lantano, A.Postigo, Chem.-Eur.J.20(2014)16806-16829; (b) J.Charpentier, N.Früh, A.Togni, Chem.Rev.115(2015)650-682. |
| [18] |
C. Zhang, Org.Biomol.Chem. 12 (2014) 6580-6589. DOI:10.1039/C4OB00671B |
| [19] |
M. Shang, S.Z. Sun, H.L. Wang, et al., Angew.Chem.Int.Ed. 53 (2014) 10439-10442. DOI:10.1002/anie.201404822 |
| [20] |
(a) M.Shang, S.Z.Sun, H.X.Dai, J.Q.Yu, J.Am.Chem.Soc.136(2014)3354-3357; (b) M.Shang, H.L.Wang, S.Z.Sun, H.X.Dai, J.Q.Yu, J.Am.Chem.Soc.136(2014)11590-11593; (c) M.Shang, S.Z.Sun, H.X.Dai, J.Q.Yu, Org.Lett.16(2014)5666-5669; (d) H.L.Wang, M.Shang, S.Z.Sun, et al., Org.Lett.17(2015)1228-1231; (e) S.Z.Sun, M.Shang, H.L.Wang, et al., J.Org.Chem.80(2015)8843-8848; (f) L.L.Xu, X.Wang, B.Ma, et al., Chem.Sci.9(2018)5160-5164 and references therein. |
| [21] |
M. Shang, Q. Shao, S.Z. Sun, et al., Chem.Sci. 8 (2017) 1469-1473. DOI:10.1039/C6SC03383K |
| [22] |
M. Shang, M.M. Wang, T.G. Saint-Denis, et al., Angew.Chem.Int.Ed. 56 (2017) 5317-5321. DOI:10.1002/anie.201611287 |
| [23] |
X. Wang, Y. Xu, F. Mo, et al., J.Am.Chem.Soc. 135 (2013) 10330-10333. DOI:10.1021/ja4056239 |
| [24] |
V. Krishnamurti, S.B. Munoz, X. Ispizua-Rodriguez, et al., Chem.Commun. 54 (2018) 10574-10577. DOI:10.1039/C8CC04907F |