 2019, Vol. 30
2019, Vol. 30 


b Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda 20892, United States
The Nobel Prize in Chemistry 2016 is awarded to Jean-Pierre Sauvage, Sir J. Fraser Stoddart and Bernard L. Feringa for their development of molecular machines that are analogues of mechanical devices with movable parts [1]. The intramolecular movements of molecular machines can be summarized into two kinds: linear or rotary. For example, most of the molecular shuttles [2] and muscles [3] are based on the linear intramolecular movements of mechanically interlocked architectures. Synthetic walkers [4] are based on the linear movements of uninterlocked dynamic systems. The molecular switches [5], tweezers [6] and rotors [7] are usually involved in conformational changes by rotation around a single bond, a double bond (isomerization) or a transition metal in organometallic systems.
The rotation-caused conformational change in biologic systems plays a key role in organisms and regulates enzyme activity for a wide variety of biochemical pathways [8]. In supramolecular chemistry, the use of conformational changes enables chemists to control molecular function by external stimuli, to transduce chemical signals, and to achieve chemical feedback regulation [9]. As analogues of calixarenes, pillararenes [10] are made up of hydroquinone units linked by methylene (-CH2-) bridges at the para-positions, forming an unique symmetrical pillar architecture. They have a relatively rigid core and multiple flexible arms which makes them ideal candidates to build up molecular machinery based on conformational changes. In this paper, we found that a fully triazole-substituted pillar[5]arene can work as a molecula excavator grapple grasps a PF6- anion through the significant structure change from pillar into conical driven by F-.
In our previous work, a non-symmetric pillar[5]arene-based receptor containing five triazole anion-binding sites on one side was designed and synthesized (Fig. S7 in Supporting information). It has high affinity and selectivity for the fluoride anion [10h]. The recognition is based on the hydrogen-bond interactions between the five triazole hydrogen atoms and the anions, which results in the conformational change from the pillar architecture into the conical one. To further investigate the guest-induced structure changes in pillararene platforms, we synthesized another symmetric receptor 4 (Scheme 1) containing ten triazole anion-binding sites. By condensation of 1 and paraformaldehyde with boron trifluoride diethyl etherate [BF3·O(C2H5)2], 2 with ten alkynyl groups was prepared (17%). Compound 3 was obtained by the treatment of 2-bromomethyl naphthalene with sodium azide in acetone/water (10:1) at 50 ℃. The reaction of 2 with 3 in the presence of CuSO4·5H2O and sodium ascorbate provided the fully triazole-substituted pillar[5]arene 4 (75%).

|
Download:
|
| Scheme 1. Synthesis and the single crystal structure of 4. | |
{kind=link}
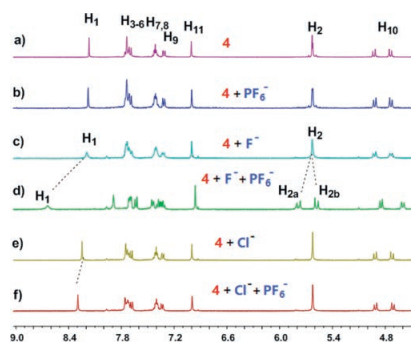
At first, we studied the binding of 4 to halide anions through 1H NMR spectra (Fig. 1). Upon the addition of 24 equiv. spherical F-, Cl-, Br- and I-, the signals related to H11 on the benzene rings and H10 on the methylene bridges of 4 showed upfield shifts slightly, which should be ascribed to the anion-induced through-bond shielding effect. As the best hydrogen bond donor on 4, triazole protons H1 shifted downfield significantly (Δδ = +0.37, +0.69, +0.44 and + 0.11 ppm) for F-, Cl-, Br- and I-, respectively, which also proved the fast exchange rate of their complexation. Interestingly, signals of H2 changed differently (the rectangle in Fig. 1). After the addition of Cl- and Br-, H2 split from a singlet into two doublets with different coupling constants (Figs. 1c and d). This indicated that the rotation of H2 was suppressed and was slow on the NMR time scale. However, H2 still was a singlet after the addition of 24 equiv. I- (Fig. 1e). For F-, part of H2 was still a singlet (H2' in Fig. 1b), part of H2 changed to a set of doublets weirdly (H2 in Fig. 1b). We speculate the reason is that, for Cl- and Br-, the complexation driven by cooperative quintuplicate hydrogen bonds changed the conformation of 4 from pillar into conical (see the sketch in Figs. 1c and d), which slowed down the rotation rate of H2 and changed its pattern to be similar with H10. Due to the largest size and lowest electronegativity of I-, the conformation of 4⊃I- did not change that much (see the sketch in Fig. 1e). As a result of the highest electronegativity and smallest size, F- might form hydrogen bond with only part of H1 (not five) without remarkable conformational change of the host structure (the left sketch of 4'⊃F- showing in Fig. 1b), which retained the singlet for H2'. The other part of 4⊃F- changed from pillar into conical, which showed the two doublets for H2 (the right sketch of Fig. 1b). To fully explore the interaction modes of 4 and halides, 1H NMR titration experiments were carried out (Figs. S8–S19 in Supporting information). During the addition of TBAF, the two doublets of H2 appeared and the integrations were increased gradually (see the rectangle in Fig. S8). Because of the balance between the pillar (4'⊃F-) and conical structures (4⊃F-), the peaks of 4 changed somehow from sharp to broad during the addition process, especially for H1 (Fig. S8). During the addition of Cl- and Br-, the H2 changed form a singlet to two doublets and the coupling constants were getting larger, indicating the rotation of H2 was suppressed gradually (see the rectangles in Figs. S11 and S14). The stoichiometry of the complexation between 4 and halide anions were all determined to be 1:1 instead of 1:2 by using mole ratio plots (Figs. S9, S12, S15 and S18). Considering the electrostatic repulsion between two anions, it is reasonable that the binding sites on two sides of 4 are too close to bind two halides at the same time. The association constants (Ka) of the complexes between 4 and halide anions were determined (106 ± 7, 262 ±16, 108 ± 17, 18 ± 4 L/mol, respectively) for F-, Cl-, Br- and I- by fitting the chemical shift changes of H1 as a function of the initial concentration.

|
Download:
|
| Fig. 1. Partial 1H NMR spectra (400 MHz, acetone-d6, 298 K): (a) 4 (1.00 mmol/L); (b) 4 (1.00 mmol/L) and TBAF (24.0 mmol/L); (c) 4 (1.00 mmol/L) and TBACl (24.0 mmol/ L); (d) 4 (1.00 mmol/L) and TBABr (24.0 mmol/L); (e) 4 (1.00 mmol/L) and TBAI (24.0 mmol/L). | |
{kind=link}
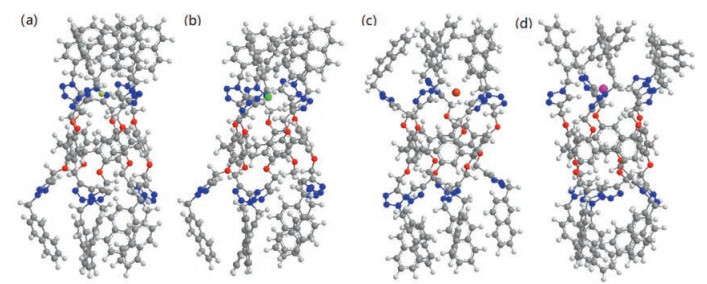
We also get the single crystal of 4 suitable for X-ray analysis by slow evaporation of a solution of 4 in a dichloromethane/ acetonitrile mixture. The resulting crystal structure (Fig. S26 in Supporting information) shows that a dichloromethane molecule can be included in the cavity of 4, demonstrating that the cavity size of 4 is enough for the inclusion of a halide anion. A hydrogen bond (B in Fig. S26) is formed between a chlorine atom of this dichloromethane molecule and a H10 hydrogen atom. Furthermore, it can be found that two intramolecular hydrogen bonds (C and D in Fig. S26) are formed between triazole hydrogen atoms and triazole nitrogen atoms in the crystal structure. This shows that triazole C-H groups can work as hydrogen bonding donors. Unfortunately, we can not obtain the X-ray crystal structure of a host–guest complex of receptor 4 and an anion. Therefore we investigated the energyminimized structures of 4⊃X (X = F-, Cl-, Br- and I-), which indicated that 4 tend to form 1:1 complexes with halide anions (Fig. 2). Five triazole hydrogens on one side of 4 wrap up one halide anion and interact with it via hydrogen bonds, while the other side of 4 can not bond another halide anion due to the cyclic structure of the receptor changed somehowfrom pillar to conical. Therefore, 1:1 complexes (instead of 1:2) between 4 and the halide anions were obtained. These informations are in good agreement with the above 1H NMR titration experiments. Due to the discrepancy in size (The diameters of F-, Cl-, Br- and I- are 1.33, 1.81, 1.96 and 2.20 Å, respectively) and electronegativity between these halide anions, the conformation of 4 changeddifferently when complexed with F-, Cl-, Br- and I-. It showed that for F- (Fig. 2a) or Cl- (Fig. 2b), the binding side was inclined to converge, while the other side was inclined to diverge. And the structure changed more notably 4⊃F- than that of 4⊃Cl-, so the free side of 4⊃F- should be with larger space for another larger guest.

|
Download:
|
| Fig. 2. Molecular models of the structures for halide complexation with 4 based on the method of B3LYP/6-31 G(D): (a) 4⊃F-; (b) 4⊃Cl-; (c) 4⊃Br-; (d) 4⊃ I-. | |
{kind=link}
As illustrated above, there are still free binding sites on 4⊃X-, the complex might be used as a host for another suitable anion. To minimize the electrostatic repulsion, the large size PF6- with low charge density (Fig. S27 in Supporting information) was selected as the other guest for 4⊃X-. As shown in Fig. 3b, when excess TBAPF6 (24 equiv.) was added into the solution of 4, no chemical shifts were observed. Indicating that the PF6- could not interact with 4 itself. However, significant chemical shift changes of 4 were observed upon the addition of TBAPF6 (24.0 mmol/L) to a solution comprised of equimolar amounts of 4 and TBAF (1.00 mmol/L). The peak for the protons H1 shifted downfield from 8.19 to 8.64 ppm. The proton signals of the methylene H2 split into two doublets (Fig. 3d). Similarly, PF6- can be triggered to interact with 4 in the presence of an equivalent amount of Cl- (Fig. 3e). The signal of the protons H1 on triazole rings showed a slight downfield shift from 8.25 ppm to 8.30 ppm. These observations indicated that the triazole rings on the free side of the pillar[5]arene-based receptor 4 can provide multiple recognition sites cooperatively for PF6- when complexed with F- or Cl-.

|
Download:
|
| Fig. 3. Partial 1H NMR spectra (400 MHz, acetone-d6, 298 K): (a) 4 (1.00 mmol/L); (b) 4 (1.00 mmol/L) and TBAPF6 (24.0 mmol/L); (c) 4 (1.00 mmol/L) and TBAF (1.00 mmol/L); (d) 4 (1.00 mmol/L), TBAF (1.00 mmol/L) and TBAPF6 (24.0 mmol/L); (e) 4 (1.00 mmol/L) and TBACl (1.00 mmol/L); (f) 4 (1.00 mmol/L), TBACl (1.00 mmol/L) and TBAPF6 (24.0 mmol/L). | |
{kind=link}
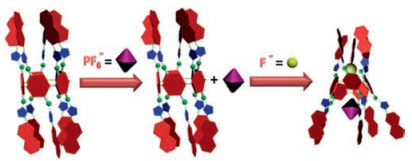
The association constants between PF6- and 4⊃F- or 4⊃Cl- were calculated to be 71.6 ± 3.8 and 29.5 ± 5.2 L/mol respectively, by conducting 1H NMR titration experiments in acetone-d6 (Figs. S20-S23 in Supporting information). No chemical shift changes related to the protons of 4 were observed upon addition of PF6- in the presence of Br- or I- (Figs. S24 and S25 in Supporting information). Therefore, similar with the molecular tweezers driven by the rotations around a single bond, the symmetric pillar[5]arene 4 containing ten triazole anion binding sites can work as a molecular grapple to grasp a PF6- anion by the F- controlled rotation of the repeating units around the methylene bridges as shown in Fig. 4.

|
Download:
|
| Fig. 4. Cartoon representation of the molecular grapple 4 grasps the PF6- anion by the addition of F-. | |
{kind=link}
In conclusion, a symmetric pillararene-based molecule 4 containing ten triazole anion-binding sites was synthesized. It showed remarkable ability to bind the halide anions and formed 1:1 complexes. This was caused by the cooperative multivalent hydrogen bonds between 4 and the halide anions. The conformational change of complexes 4⊃F- or 4⊃Cl- affords the capture of the large-size anion PF6- on the other side. Therefore, this fully triazole substituted pillar[5]arene 4 can work as a molecular grapple selectively grasps a PF6- anion by the control of F-. To the best of our knowledge, due to its large size and lowelectronegativity, almost no anion receptor that is selectiveforPF6- hasbeen reportedup tonow. Because of the special structure with a relatively rigid core and certain flexible functionalized arms, we believe that pillararenewill be an ideal platform to build up molecular machinery based on the controllable conformation changes.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21572042 and 21773052), the Program for Innovative Research Team in Chinese University (No. IRT 1231), the Zhejiang Provincial Natural Science Foundation of China (Nos. LZ16B020002 and LQ17B040002), and the Pandeng Plan Foundation of Hangzhou Normal University for Youth Scholars of Materials, Chemistry and Chemical Engineering.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.01.018.
| [1] |
(a) B.L.Feringa, Acc.Chem.Res.34(2001)504-513; (b) B.Champin, P.Mobian, J.P.Sauvage, Chem.Soc.Rev.36(2007)358-366; (c) A.Coskun, M.Banaszak, R.D.Astumian, J.F.Stoddart, B.A.Grzybowski, Chem.Soc.Rev.41(2012)19-30. |
| [2] |
(a) K.Zhu, C.A.O'Keefe, V.N.Vukotic, R.W.Schurko, S.J.Loeb, Nat.Chem.7(2015)514-519; (b) G.Yu, D.Wu, Y.Li, et al., Chem.Sci.7(2016)3017-3024; (c) K.Zhu, G.Baggi, S.J.Loeb, Nat.Chem.10(2018)625-630. |
| [3] |
(a) Z.Zhang, C.Han, G.Yu, F.Huang, Chem.Sci.3(2012)3026-3031; (b) K.Zhu, V.N.Vukotic, S.J.Loeb, Angew.Chem.Int.Ed.51(2012)2168-2172; (c) C.J.Bruns, J.F.Stoddart, Acc.Chem.Res.47(2014)2186-2199; (d) L.Gao, Z.Zhang, B.Zheng, F.Huang, Polym.Chem.5(2014)5734-5739; (e) P.Wang, Z.Gao, M.Yuan, J.Zhu, F.Wang, Polym.Chem.7(2016)3664-3668. |
| [4] |
(a) M.von Delius, E.M.Geertsema, D.A.Leigh, Nat.Chem.2(2010)96-101; (b) J.E.Beves, V.Blanco, B.A.Blight, et al., J.Am.Chem.Soc.136(2014)2094-2100; (c) C.J.Martin, A.T.L.Lee, R.W.Adams, D.A.Leigh, J.Am.Chem.Soc.139(2017)11998-12002. |
| [5] |
(a) S.Fukuzumi, K.Ohkubo, Y.Kawashima, et al., J.Am.Chem.Soc.133(2011)15938-15941; (b) Q.Chen, J.Sun, P.Li, et al., J.Am.Chem.Soc.138(2016)14242-14245; (c) M.Cheng, J.Zhang, X.Ren, et al., Chem.Commun.53(2017)11838-11841. |
| [6] |
(a) M.Hardouin-Lerouge, P.Hudhomme, M.Salle, Chem.Soc.Rev.40(2011)30-43; (b) T.Fu, Y.Han, L.Ao, F.Wang, Organometallics 35(2016)2850-2853; (c) Z.Gao, Y.Han, S.Chen, et al., ACS Macro Lett.6(2017)541-545. |
| [7] |
(a) D.A.Leigh, J.K.Y.Wong, F.Dehez, F.Zerbetto, Nature 424(2003)174-179; (b) A.Comotti, S.Bracco, T.Ben, S.Qiu, P.Sozzani, Angew.Chem.Int.Ed.53(2014)1043-1047; (c) J.Chen, F.K.C.Leung, M.C.A.Stuart, et al., Nat.Chem.10(2018)132-138. |
| [8] |
J. Guo, H.X. Zhou, Chem.Rev. 116 (2016) 6503-6515. DOI:10.1021/acs.chemrev.5b00590 |
| [9] |
(a) J.L.Sessler, E.Tomat, V.M.Lynch, J.Am.Chem.Soc.128(2006)4184-4185; (b) E.R.Kay, D.A.Leigh, F.Zerbetto, Angew.Chem.Int.Ed.46(2007)72-191. |
| [10] |
(a) T.Ogoshi, S.Kanai, S.Fujinami, T.A.Yamagishi, Y.Nakamoto, J.Am.Chem.Soc.130(2008)5022-5023; (b) D.R.Cao, Y.H.Kou, J.Q.Liang, et al., Angew.Chem.Int.Ed.48(2009)9721-9723; (c) Z.Zhang, B.Xia, C.Han, Y.Yu, F.Huang, Org.Lett.19(2010)4360-4363; (d) C.Li, L.Zhao, J.Li, et al., Chem.Commun.46(2010)9016-9018; (e) Z.Zhang, Y.Luo, J.Chen, et al., Angew.Chem.Int.Ed.50(2011)1397-1401; (f) N.L.Strutt, R.S.Forgan, J.M.Spruell, Y.Y.Botros, J.F.Stoddart, J.Am.Chem.Soc.133(2011)5668-5671; (g) W.Si, L.Chen, X.B.Hu, et al., Angew.Chem.Int.Ed.50(2011)12564-12568; (h) G.Yu, Z.Zhang, C.Han, et al., Chem.Commun.48(2012)2958-2960; (i) S.Pan, M.Ni, B.Mu, et al., Adv.Funct.Mater.25(2015)3571-3580; (j) X.Chi, G.Yu, L.Shao, J.Chen, F.Huang, J.Am.Chem.Soc.138(2016)3168-3174; (k) M.Ni, N.Zhang, W.Xia, et al., J.Am.Chem.Soc.138(2016)1386643-1386649; (l) J.C.Gui, Z.Q.Yan, Y.Peng, et al., Chin.Chem.Lett.27(2016)1017-1021; (m) B.Shi, K.Jie, Y.Zhou, et al., J.Am.Chem.Soc.138(2016)80-83; (n) X.Wu, Y.Zhang, Y.Lu, et al., J.Mater.Chem.B 5(2017)3483-3487; (o) K.Jie, Y.Zhou, E.Li, et al., J.Am.Chem.Soc.139(2017)15320-15323; (p) G.Ping, Y.Wang, L.Shen, et al., Chem.Commun.53(2017)7381-7384; (q) C.B.Yin, Y.Han, G.F.Huo, J.Sun, C.G.Yan, Chin.Chem.Lett.28(2017)431-436; (r) X.L.Wu, Y.Chen, W.J.Hu, et al., Org.Biomol.Chem.15(2017)4897-4900; (s) K.Jie, Y.Zhou, E.Li, et al., J.Am.Chem.Soc.140(2018)3190-3193; (t) Y.X.Wang, Q.F.Zhou, L.J.Chen, et al., Chem.Commun.54(2018)2224-2227; (u) L.Ma, S.Wang, C.Li, et al., Chem.Commun.54(2018)2048-2405; (v) Y.M.Zhang, W.Zhu, W.J.Qu, et al., Chem.Commun.54(2018)4549-4552; (w) Q.Zhao, Y.Liu, Chem.Commun.54(2018)7362-7365. |