 2019, Vol. 30
2019, Vol. 30 


In the past decades, halogen bonding has been established as a robust non-covalent force in supramolecular engineering [1]. Due to its orthogonality, in recent years the applications of halogen bonding in materials design [2], chemical biology [3], drug discovery [4], and catalysis [5] have also been extensively investigated. Macrocyclic molecules have played a key role in molecular recognition and self-assembly due to their high binding selectivity and strength. Typically, macrocyclic molecules have been generated through the formation of conventional or dynamic covalent bonds [6, 7]. However, examples of using weak noncovalent interaction(s) to construct supramolecular macrocyclic structures are very limited [8-10], even though they are potentially useful as more adaptable artificial receptors for molecular recognition.
It has been demonstrated that the backbones of oligomeric aromatic amides can be induced by intramolecular hydrogen bonding into folded or helical conformations [11], which have been utilized to promote the formation of a variety of macrocyclic compounds [12]. Previously, we reported that orthogonal halogen bonding is robust in inducing short hydrogen bonded aromatic amide foldamers into supramolecular helical structures of varying complexity [13]. We herein describe that halogen bonding can be used to hold folded molecules to form dimeric macrocycles in the solid state. Depending on the condition of crystallization, one molecule also produces a unique highly compact supramolecular double helix with opposite component helicity.
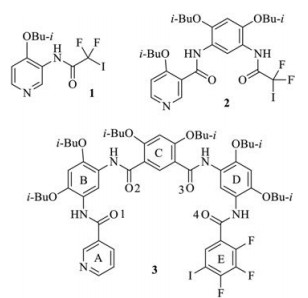
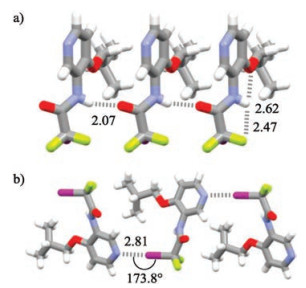
A number of compounds that contain halogen bonding donor and acceptor at the ends were prepared and the crystal structures of compounds 1-3 were obtained. The synthesis and characterization of the three compounds are provided in the Supporting information. For all compounds, pyridine N atom was chosen as halogen bonding acceptor [1a–c], while the iso-butoxyl group was introduced to the aromatic amide backbones to increase the crystallinity [14]. Compound 1 was designed as model to test the formation of intermolecular CF2I…N(py) halogen bonding as well as its orthogonality. As expected, 1 formed a strong five-membered N—H…OBu-i hydrogen bond (N…H distance: 2.62 Å) and one of the F atoms was also engaged in a five-membered N—H…F hydrogen bond (F…H distance: 2.47 ) (Fig. 2a). The amide group distorted considerably (39.8°) from the benzene ring to enable the formation of strong intermolecular N—H…O=C hydrogen bond (O…H distance: 2.07 ), even though no aromatic stacking occurred. All the three hydrogen bonds rigidified the molecule and caused the iodine atom to be located to the opposite side of the pyridine N atom, and the neighboring molecules formed strong I…N halogen bonding (I…N distance: 2.81 , sum of the van der Waals radium of I and N: 3.53 ), which held the molecules together to produce zigzag supramolecular polymeric chain. The C—I…N angle is 173.8°. Both data supported the important electrostatic attraction between the two atoms. The fact that the carbonyl O atom did not form halogen bonding with the I atom clearly reflects the orthogonality of the intermolecular I…N halogen bonding.

|
Download:
|
| Fig. 1. The structures of compounds 1–3. | |
{kind=link}

|
Download:
|
| Fig. 2. Crystal structure of compound 1 (CCDC No. 1881067), highlighting the intermolecular (a) N-H…O=C hydrogen bonding and (b) I…N halogen bonding. The crystals were grown by evaporation of the solution in dichloromethane and methanol (5:1). | |
{kind=link}
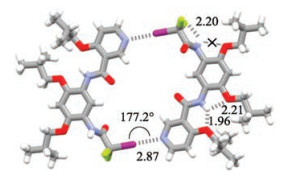
The crystal structure of compound 2 is shown in Fig. 3. The NH on the pyridine ring side was engaged in strong intramolecular three-center N—H…O hydrogen bonding (O…H distance: 1.96 and 2.21 ). In contrast, the amide group on the other side distorted greatly (63.3°) from the benzene ring to which it was connected and thus did not form intramolecular N—H…O hydrogen bonding with the adjacent ether O atom. Instead, this amide formed a stable intramolecular five-membered N—H…F hydrogen bonding (F…H distance: 2.20 ) with one of the two F atoms. These hydrogen bonds and the great distortion of the second amide group forced the— C—I bond to point to the direction parallel with that of the dipole of the pyridine ring. As a result, two molecules formed a dimeric macrocycle which was stabilized by a pair of strong I…N halogen bonds (I…N distance: 2.87 , C—I…N angle: 177.2°. It is well-established that intramolecular five-membered N—H…O hydrogen bonding of aromatic amide is highly favored as compared with intermolecular strong hydrogen bonding of the amide group [11a, b], whereas fluorine connected to the aliphatic chain has been revealed to be very weak hydrogen bonding acceptor [15]. The formation of the macrocycle involved the engagement of two intermolecular I…N halogen bonds, which caused the breaking of two strong intramolecular N—H…O hydrogen bonds. Although other factors such as aromatic stacking, which did occur, might facilitate the formation of the halogen bonding, this result also demonstrated the great strength of halogen bonding in competing with conventional hydrogen bonding, and the intramolecular hydrogen bonding might also promote the halogen bonding with each other. The dimeric macrocycle also gave rise to a rectangular cavity (approximately 0.4 nm × 1.1 nm), which was seized by the iso-butyl group and thus no solvent encapsulation was observed. The fact that compound 1 did not form macrocyclic system implied that the importance of the hydrogen bonded rigidified aromatic backbone in facilitating the formation of the halogen bonded macrocycle.

|
Download:
|
| Fig. 3. Crystal structure of compound 2 (CCDC No.1881070) showing the formation of halogen bonded macrocycle. The crystal was grown from the solution of dichloromethane and methanol (5:1). | |
{kind=link}
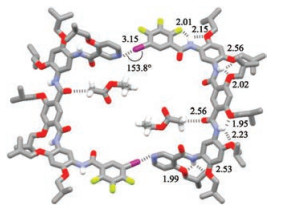
Crystals of compound 3 were first grown from the solution of dichloromethane, methanol, ethyl acetate and dimethyl sulfoxide (3:1:1:0.05). The crystal structure revealed the formation of another dimeric macrocycle (Fig. 4), which was also induced by two intermolecular I…N halogen bonds. The I…N distance (3.15 ) was considerably larger than that of 2, which was consistent with a smaller C—I…N angle (153.8°), reflecting the decreased strength of this halogen bonding. Despite of the decreased strength, the two halogen bonds still held two molecules to form a chair-styled macrocycle. All the four amide units were engaged in three-center hydrogen bonding. The related bond distances are provided in Fig. 4. Two amides (1 and 3, see Fig. 1 for numbering) distorted substantially from the attached pyridine or benzene rings. As a result, the whole aromatic backbone gave rise to a saddle-like conformation and the halogen-bonded macrocycle produced a rectangular cavity (approximately 1.0 nm × 1.6 nm). One such halogen bonded macrocycle entrapped two ethyl acetate molecules through intermolecular =(O=C)C—H…O=C (amide 2) hydrogen bonding. The entrapped ethyl acetate also formed another C=O…H—C hydrogen bonding with one of the iso-butyl groups of another halogen bonded macrocycle, which might also partially account for the considerable distortion of the aromatic backbone. Molecular modelling revealed that when the aromatic backbone of 3 adopted a planar folded conformation, the dipole of thepyridine ring and the— C—Ibondofbenzene ringEwouldform an intersection angle of approximately 120°, which is highly unfavorable for the formation of a bimolecular macrocycle. It was reasonable to propose that the saddle-like distorted conformation of 3 in the crystal structure was formed to allow the two peripheral aromatic rings to form two intermolecular halogen bonds, which should be the strongest intermolecular interactions for such a 5-mer foldamer.

|
Download:
|
| Fig. 4. Crystal structure of compound 3 (CCDC No.1881068) showing the formation of halogen bonded supramolecular macrocycle. The crystal was grown from the solution of dichloromethane, ethyl acetate, methanol and dimethylsulfoxide (3:1:1:0.05). Hydrogen atoms on carbons were omitted for clarity. | |
{kind=link}
Previous study for the shorter 3- and 4-mer analogues of 3 revealed the formation of halogen bonded supramolecular helix [13]. The fact that 3 formed halogen bonded macrocycle through distorting its aromatic backbone raised the role of the entrapped ethyl acetate molecules in inducing the macrocyclic structure. Thus, single crystals of 3 suitable for X-ray diffraction analysis were further grown from dichloromethane, methanol and dimethyl sulfoxide (3:1:0.05). The crystal structure in this case did not exhibit any solvent entrapment, and the intramolecular fivemembered C—H…O hydrogen bonding of amide 2 was broken due to large distortion of the second amide from benzene ring B (57.8°) (Fig. 1 for labeling). Moreover, the aromatic backbone occurred in both P and M crescent helicity, as defined along the backbone from pyridine ring A to benzene ring E. Surprisingly, backbones with opposite helicity were linked alternately through intermolecular I…O=C (amide 1) halogen bonding (Fig. 5a), which led to the formation of a unique supramolecular helix with the components adopting opposite helicity (Fig. 5b). The I…O distance was 3.20 and the C—I…O angle was 160.4°, reflecting a relatively weak electrostatic attraction. However, the fact that no I…N halogen bonding was formed as the former case indicated that this halogen bonding was stronger than the "designed" I…N halogen bonding, which might be attributed to complicated steric effect and other intramolecular and/or intermolecular interactions. The pyridine N atom formed a weak intermolecular N…H—C (N…H distance: 2.63 ) hydrogen bonding, which should stabilize the extended aggregation of the supramolecular helices. The neighboring single supramolecular helices further stacked to form new double helix (Fig. 5b), which was stabilized by cross-chain stacking between the P components of one chain and the M components of another one. That is, the neighboring supramolecul helices aggregated alternately through opposite arrangement. This stacking extended into two-dimensional space to afford a mono-layered helix array.

|
Download:
|
| Fig. 5. (a) Conformational enantiomers in the crystals of compound 3 (CCDC No. 1881069) grown by evaporation of the solution in dichloromethane, methanol and dimethyl sulfoxide (3:1:0.05). (b) Double helix formed by two supramolecular helical polymers composed of alternately connected P (light blue and pink) and M (blue and red) folded components. The helicity was defined along the backbone from pyridine ring A to benzene ring E. The iso-butoxyl chain and hydrogen atoms were omitted for clarity. | |
{kind=link}
In conclusion, we have demonstrated that halogen bonding is a useful non-covalent force for the formation of supramolecular macrocycles when the molecular components adopt a crescent or folded conformation. For bimolecular macrocycles, the high directionality of halogen bonding requires the halogen bonding donor and acceptor to arrange in roughly parallel manner. To meet this requirement, the backbone of the molecular component may distort considerably by sacrificing other relatively weaker noncovalent forces. In principle, by rational design, two or more identical or different molecular components may be combined to form macrocycles with varying cavity. In the future, we will investigate this possibility and related molecular encapsulation.
AcknowledgmentWe thank the National Natural Science Foundation of China (Nos. 21772026 and 21432004) for financial support.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.02.010.
| [1] |
(a) A.Priimagi, G.Cavallo, P.Metrangolo, G.Resnati, Acc.Chem.Res.46(2013)2686-2695; (b) L.C.Gilday, S.W.Robinson, T.A.Barendt, et al., Chem.Rev.115(2015)7118-7195; (c) P.Politzer, P.Lane, M.C.Concha, Y.Ma, J.S.Murray, J.Mol.Model.13(2007)305-311; (d) K.Rissanen, CrystEngComm 10(2008)1107-1113; (e) A.Mukherjee, S.Tothadi, G.R.Desiraju, Acc.Chem.Res.47(2014)2514-2524; (f) T.M.Beale, M.G.Chudzinski, M.G.Sarwar, M.S.Taylor, Chem.Soc.Rev.42(2013)1667-1680; (g) H.Wang, W.Wang, W.J.Jin, Chem.Rev.116(2016)5072-5104; (h) X.Pang, W.J.Jin, Top.Curr.Chem.359(2015)115-146; (i) C.Z.Liu, H.Wang, D.W.Zhang, X.Zhao, Z.T.Li, Chin.J.Org.Chem.39(2019)28-37. |
| [2] |
(a) J.Li, Y.H.Hu, C.W.Ge, H.G.Gong, X.K.Gao, Chin.Chem.Lett.29(2018)423-428; (b) M.Wang, C.Cheng, J.Song, et al., Chin.J.Chem.36(2018)698-707; (c) A.Forni, E.Lucenti, C.Botta, E.Cariati, J.Mater.Chem.C 6(2018)4603-4626; (d) M.Saccone, G.Cavallo, P.Metrangolo, G.Resnati, A.Priimagi, Top.Curr.Chem.359(2015)147-166; (e) D.Yan, H.Yang, Q.Meng, H.Lin, M.Wei, Adv.Funct.Mater.24(2014)587-594; (f) G.Fan, D.Yan, Adv.Optical Mater.4(2016)2139-2147; (g) D.Yan, D.K.BuÅ263;ar, A.Delori, et al., Chem.-Eur.J.19(2013)8213-8219. |
| [3] |
(a) A.M.Montana, ChemistrySelect 2(2017)9094-9112; (b) M.R.Scholfield, M.C.Ford, A.C.C.Carlsson, et al., Biochemistry 56(2017)2794-2802; (c) R.Wilcken, M.O.Zimmermann, A.Lange, A.C.Joerger, F.M.Boeckler, J.Med.Chem.56(2013)1363-1388. |
| [4] |
(a) L.Mendez, G.Henriquez, S.Sirimulla, M.Narayan, Molecules 22(2017)1397; (b) M.C.Ford, P.S.Ho, P.J.Med.Chem.59(2016)1655-1670; (c) Y.Lu, Y.Liu, Z.Xu, et al., Exp.Opin.Drug Discov.7(2012)375-383. |
| [5] |
(a) G.Bergamaschi, L.Lascialfari, A.Pizzi, et al., Chem.Commun.54(2018)10718-10721; (b) Y.C.Chan, Y.Y.Yeung, Angew.Chem.Int.Ed.57(2018)3483-3487; (c) Y.Wang, J.Wang, G.X.Li, G.He, G.Chen, Org.Lett.19(19)(2017)1442-1445; (d) S.Jiang, L.Zhang, D.Cui, et al., Sci.Rep.6(2016)34750; (e) X.Sun, W.Wang, J.Ma, S.Yu, Acta Chim.Sinica 75(2017)115-118. |
| [6] |
(a) R.Peng, Y.Xu, Q.Cao, Chin.Chem.Lett.29(2018)1465-1474; (b) S.Zhang, L.Zhao, Acc.Chem.Res.51(2018)2535-2545; (c) C.W.Sathiyajith, R.R.Shaikh, Q.Han, et al., Chem.Commun.53(2017)677-696; (d) Y.D.Yang, J.L.Sessler, H.Y.Gong, Chem.Commun.53(2017)9684-9696; (e) Z.Li, J.Liang, W.Xue, et al., Supramol.Chem.26(2014)54-65; (f) W.Q.Ong, H.Zeng, J.Inclus.Phenom.Macrocycl.Chem.76(2013)1-11; (g) Y.Lu, D.D.Liang, Z.D.Fu, Q.H.Guo, M.X.Wang, Chin.J.Chem.36(2018)630-634; (h) G.Ji, S.Zhang, S.C.K.Hau, L.Zhao, Chin.J.Chem.35(2017)1824-1828; (i) T.Xiao, W.Zhong, L.Zhou, X.Y.Hu, L.Wang, et al., Chin.Chem.Lett.30(2019)31-36. |
| [7] |
(a) Y.Jin, Q.Wang, P.Taynton, W.Zhang, Acc.Chem.Res.47(2014)1575-1586; (b) L.Yuan, Y.Han, T.Tao, H.Phan, C.Chi, Angew.Chem.Int.Ed.57(2018)9023-9027; (c) Y.Zhang, X.Zheng, N.Cao, C.Yang, H.Li, Org.Lett.20(2018)2356-2359; (d) X.He, Y.Xue, C.C.Li, et al., Chem.Sci.9(2018)1481-1487; (e) S.Yang, Z.Luan, C.Gao, J.Yu, D.Qu, Sci.China Chem.61(2018)306-310; (f) B.Yang, S.B.Yu, H.Wang, D.W.Zhang, Z.T.Li, Chem.-Asian J.13(2018)1312-1317; (g) X.F.Li, S.B.Yu, B.Yang, et al., Sci.China Chem.61(2018)830-835. |
| [8] |
(a) C.M.Drain, K.C.Russell, J.M.Lehn, Chem.Commun.(1996)337-338; (b) L.J.Marshall, J.de Mendoza, Org.Lett.15(2013)1548-1551. |
| [9] |
W.K. Wang, Y.Y. Chen, H. Wang, et al., Chem.-Asian J. 9 (2014) 1039-1044. DOI:10.1002/asia.v9.4 |
| [10] |
P.M.J. Szell, A. Siiskonen, L. Catalano, et al., New J.Chem. 42 (2018) 10467-10471. DOI:10.1039/C8NJ00759D |
| [11] |
(a) B.Gong, Chem.-Eur.J.7(2001)4336-4342; (b) I.Huc, Eur.J.Org.Chem.(2004)17-29; (c) Z.T.Li, J.L.Hou, C.Li, Acc.Chem.Res.41(2008)1343-1353; (d) B.Gong, Acc.Chem.Res.41(2008)1376-1386; (e) A.Roy, P.Prabhakaran, P.K.Baruah, G.J.Sanjayan, Chem.Commun.47(2011)11593-11611; (f) Q.Gan, Y.Wang, H.Jiang, Curr.Org.Chem.5(2011)1293-1301; (g) D.W.Zhang, X.Zhao, J.L.Hou, Z.T.Li, Chem.Rev.112(2012)5271-5316; (h) D.W.Zhang, X.Zhao, Z.T.Li, Acc.Chem.Res.47(2014)1961-1970; (i) Y.Huo, H.Zeng, Acc.Chem.Res.49(2016)922-930; (j) C.Z.Liu, M.Yan, H.Wang, D.W.Zhang, Z.T.Li, ACS Omega 3(2018)5165-5176; (k) G.Sun, C.Nie, X.Zhao, Z.Li, Chin.J.Org.Chem.37(2017)1757-1763; (l) L.Yang, W.Zhao, Y.K.Che, Y.Wang, H.Jiang, Chin.Chem.Lett.28(2017)1659-1662; (m) D.W.Zhang, H.Wang, Z.T.Li, Macromol.Rapid Commun.38(2017)1700179. |
| [12] |
(a) L.Yuan, W.Feng, K.Yamato, et al., J.Am.Chem.Soc.126(2004)11120-11121; (b) H.Jiang, J.M.LÅ233;ger, P.Guionneau, I.Huc, Org.Lett.6(2004)2985-2988; (c) F.Li, Q.Gan, L.Xue, Z.M.Wang, H.Jiang, Tetrahedron Lett.50(2009)2367-2369; (d) H.Fu, Y.Liu, H.Zeng, Chem.Commun.49(2013)4127-4144; (e) Y.He, M.Xu, R.Gao, et al., Angew.Chem.Int.Ed.53(2014)11834-11839; (f) B.Qin, X.Chen, X.Fang, et al., Org.Lett.10(2008)5127-5130; (g) P.Xin, L.Zhang, P.Su, J.L.Hou, Z.T.Li, Chem.Commun.51(2015)4819-4822; (h) J.B.Lin, X.N.Xu, X.K.Jiang, Z.T.Li, J.Org.Chem.73(2008)9403-9410; (i) X.N.Xu, L.Wang, Z.T.Li, Chem.Commun.(2009)6634-6636. |
| [13] |
C.Z. Liu, S. Koppireddi, H. Wang, D.W. Zhang, Z.T. Li, Angew.Chem.Int.Ed. 58 (2019) 226-230. DOI:10.1002/anie.201811561 |
| [14] |
H. Jiang, J.M. Léger, I. Huc, J.Am.Chem.Soc. 25 (2003) 3448-3449. |
| [15] |
H.A.K. Howard, V.J. Hoy, D. O'Hagan, G.T. Smith, Tetrahedron 52 (1996) 12613-12622. DOI:10.1016/0040-4020(96)00749-1 |