 2019, Vol. 30
2019, Vol. 30 


b School of Physics and Nuclear Energy Engineering, Beihang University, Beijing 100191, China
In the past decades, polymers containing azobenzene and its derivatives (azo polymers for short) have attracted a wide range of attention owing to their unique photoresponsive properties and potential applications in areas such as holographic recording, optical switching, sensors, nonlinear optics, and photorefractive materials [1-10]. When azo polymer films are irradiated with light at an appropriate wavelength, a variety of photoresponsive variations, such as phase transition [11], chromophore orientation [12], and surface-relief-grating (SRG) formation [13, 14], are triggered by the trans-cis photoisomerization of azo chromophores [15]. Photoinduced birefringence and dichroism are caused by repeated trans-cis-trans photoisomerization and orientation of azo chromophores, which undergo an angular redistribution predominating in the direction perpendicular to the polarization plane of the actinic light [2-4]. SRG formation refers to the surface modulation on azo polymer films when irradiated with interfering laser beams [3-5, 13, 14]. Sinusoidal surface patterns with modulated depths in submicron scale can be inscribed on azo polymer films at a temperature below the glass transition temperature (Tg) of the polymers, which are erasable by heating the films to a temperature above their Tg [4, 5]. The surface patterns can also be erased by irradiation with a circularly polarized laser beam below Tg of the polymers [4, 5]. Besides the side-chain azo polymers, which have been most widely investigated, the SRG formation has also been studied by using hyperbranched azo polymers [16], main-chain azo polymers [17], polymers with conjugated main-chain [18], and others [3-5, 9, 10]. The SRG formation has been attributed to mass transfer caused by light irradiation. Different models and theories have been proposed to elucidate the mechanisms [19-26]. Although it is generally agreed that the photoinduced trans-cis isomerization of azo chromophores plays a critically important role in the process, the deep understanding of the exact mechanism is still a subject under consistent investigations.
Amorphous molecular materials, also known as molecular glasses, have recently been developed as a new type of amorphous materials to contain azo chromophores [27-31]. Although their molecular weights are much lower than those of their polymeric counterparts, the materials can exist in a stable glass state below Tg. To be used as photoresponsive media, amorphous molecular materials containing azo chromophores (azo molecular glasses) show some obvious advantages, such as well-defined chemical structures, high density of functional groups, easy purification, and lack of chain entanglements of polymers [27, 28, 30]. Meanwhile, solid thin films can be obtained by conventional spin-coating technique, which show an excellent optical quality comparable with polymeric films. Azo molecular glasses usually show a fast response to the light irradiation, which is a very attractive characteristic for optical data-storage, holographic recording, surface patterning and other applications [28, 31]. Typical types of azo molecular glasses include rigid molecules containing one or two azobenzene moieties [27, 29, 30], three-arm or star-shape molecules [32-38], linear molecules with an azo chromophore linked to a biphenylene unit through a flexible spacer [39, 40].
According to the spectral feature and isomerization behavior, azo chromophores have been classified into azobenzene type, aminoazobenzene type, and pseudo-stilbene type [15]. The pseudo-stilbene (push-pull) type azo chromophores typically contain strong electron-donating/-withdrawing substituents at 4- and 4'-positions of the azobenzene moieties. Upon the light irradiation at proper wavelengths, the push-pull type azo chromophores undergo quick trans-cis-trans isomerization cycles. Consequently, the azo chromophores and adjoining groups will be swiftly orientated in the azo polymer films to show significant birefringence and dichroism [2-4]. Due to the quick isomerization of this type of azo chromophores, SRG can also be efficiently inscribed on the polymer film surfaces [3-5, 13, 14]. However, up till now, there are few reports related to the systematic investigation on the molecular glasses containing the push-pull type azo chromophores. Although the chemical structure of azo molecular glasses can be well characterized, the relationship between the chromophoric structures and photoresponsive properties under the light excitation at different wavelengths has not be fully understood and elucidated.
In this study, a series of epoxy-based azo molecular glasses were synthesized to have the four-arm architecture, which contain four push-pull type azo chromophores at their peripheral positions. The molecules were synthesized through the ring-opening reaction between 4, 4'-methylene-bis(N, N'-diglycidyl-aniline) and N-methylaniline to yield the intermediate MDGA-AN. It was then functionalized by the azo-coupling reactions with five different diazonium salts. The photoinduced birefringence was studied by exposure of the azo molecular glass films to a linearly polarized laser beam at three different wavelengths (λex = 488, 532 and 589 nm). The SRG formation was investigated by irradiation with p, p-polarized interfering laser beams at these wavelengths. The influences of chromophoric structure and excitation wavelength on the photoresponsive behavior were investigated in details. Excellent photoresponsive properties were observed for some of the azo molecular glasses when irradiated with light at appropriate wavelengths. One of the materials with the high efficiency for the photoinduced orientation was selected for polarization holographic recording to demonstrate the potential application of the materials.
The synthetic route and the chemical structure of the azo molecular glasses are shown in Scheme 1. The intermediate (MDGA-AN) was prepared through the ring-opening reaction between 4, 4'-methylene-bis(N, N'-diglycidylaniline) (MDGA) and N-methylaniline (AN). The synthesis details and the characterization of MDGA-AN are shown in Supporting information (Figs. S1 and S2 in Supporting information). The five azo molecular glasses MDGA-AZ-A (A: NT, CN, CA, F, MO) were synthesized through azocoupling reaction between MDGA-AN and the diazonium salts with different electron-withdrawing groups. The abbreviated names of NT, CN, CA, F, MO respectively stand for the nitro, cyano, carboxyl, fluoro and methoxyl groups in the para-position. Synthetic procedure of MDGA-AZ-NT is shown below as an example.

|
Download:
|
| Scheme 1. Synthetic route and chemical structure of MDGA-AZ-A (A: NT, CN, CA, F, MO). | |
4-Nitroaniline (1.04 g, 7.56 mmol) was dissolved in a homogeneous mixture of acetic acid (12 mL), propionic acid (9 mL) and sulfuric acid (98%, 1.8 mL) at 0 ℃. Diazonium salt was prepared by adding an aqueous solution of sodium nitrite (0.73 g, 10.58 mmol in 1 mL of water) dropwise into the 4-nitroaniline solution. The mixture was stirred below 5 ℃ for 5 min and then added dropwise into a solution of MDGA-AN (1.43 g, 1.68 mmol) in N, N'-dimethylformamide (DMF, 60 mL) at 0 ℃. The reaction was carried out with stirring at 0 ℃ for 12 h. The solution was then added dropwise into plenty of water, and the precipitate was collected by filtration and vacuum-dried. The obtained solid was dissolved in THF, and the solution was filtrated and precipitated again with abundant petroleum ether. The product was collected by filtration and dried in a vacuum oven at 45 ℃ for 48 h. MDGA-AZ-CN, MDGA-AZ-CA, MDGA-AZ-F, and MDGA-AZ-MO were prepared via the same procedure as mentioned above, but respectively using the diazonium salt of 4-aminobenzonitrile, 4-aminobenzoic acid, 4-fluoroaniline and 4-methoxyaniline instead.
The successful syntheses of the azo compounds with the designed structures were confirmed by 1H NMR (Fig. S3 in Supporting inforamtion) and FT-IR (Fig. S4 in Supporting information). In the 1H NMR spectrum of the intermediate MDGA-AN, the resonance signals at δ6.78, 6.62, 6.46 correspond to the chemical shifts of the protons at meta, ortho, and para positions of the peripheral aniline moieties. After the azo-coupling reaction, the resonance signal at δ6.46 completely disappears due to the electrophilic substitution at the para position. Moreover, additional resonance signals corresponding to the introduced benzenoid ring protons appear at the lower magnetic field. The result indicates that for the azo-coupling reactions, the conversions of anilino moieties of the precursor MDGA-AN were about 100%. The assignment of the resonance signals is given in Fig. S3 and the assignment of the absorption bands of the FTIR spectra to the vibration modes of the compounds are given in the Supporting information. The thermal behavior and phase transition temperatures of MDGA-AZ-A (A: NT, CN, CA, F, MO) were characterized by the differential scanning calorimetry (DSC, Fig. S5 in Supporting information). All azo compounds obtained in this study showed typical behavior of the amorphous materials. The Tg values of the azo compounds (Table 1) are significantly higher than that of MDGA-AN (44 ℃) due to the introduction of the polar azo chromophores. The decomposition temperature (Td) values obtained from the thermal gravimetric analysis (TGA, Fig. S6 in Supporting information) are also given in Table 1. Compared with MDGA-AN, the Td values of the azo molecular glasses decline, which can be attributed to the lower thermal stability of the azo groups.
|
|
Table 1 The glass transition temperature (Tg), the decomposition temperature (Td), and λmax of MDGA-AZ-A (A: NT, CN, CA, F, MO). |

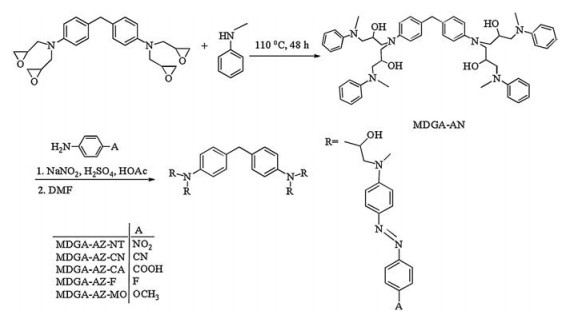
Fig. 1a shows UV–vis absorption spectra of MDGA-AZ-A (A: NT, CN, CA, F, MO) in DMF solutions. Except MDGA-AZ-MO, the azo compounds show typical spectral characteristics of the pseudostilbene type azo chromophores. The band with the strong absorption, corresponding to the π→π* transition, appears in the visible spectrum region and the weak n→π* transition band is buried in the strong π→π* transition band. Due to the resonancedonating (π-donating) effect of the methoxy group, MDGA-AZ-MO shows spectral features different to the others. Besides the π→π* transition band at 431 nm, there is another weaker absorption band in UV range (λmax = 366 nm). The λmax values of the π→π* transitions for the azo compounds are listed in Table 1. The λmax values of the azo compounds are strongly affected by the parasubstituents on the azo chromophores. As the electron-withdrawing capability increases, the λmax increases in the order of fluoro (F), carboxyl (CA), cyano (CN) and nitro (NT) substitutes. Fig. 1b shows UV–vis absorption spectra of the solid thin films of the azo molecular glasses. Except MDGA-AZ-MO, the λmax values of the solid films appear at the obviously shorter wavelengths compared with those of the corresponding DMF solutions (Table 1), which are attributed to the solvatochromic effect of DMF. The extent of the red-shift, caused by the solvatochromic effect, shows the order for substitutes F < CA < NT < CN. On the other hand, for methoxy as the para-substituent, no such solvatochromic effect is observed for MDGA-AZ-MO, which is also attributed to the π-donating effect of the methoxy group.

|
Download:
|
| Fig. 1. UV–vis spectra of the azo compounds: (a) DMF solution, (b) azo compound films. | |
The molar extinction coefficients (ε) of the azo compounds were calculated from the UV–vis spectra of their DMF solutions using the Beer-Lambert law, based on the linear correlation between the absorbance and concentration (Fig. 2). The ε values were obtained at λmax of their π→π* transition bands and at three other wavelengths (488, 532, and 589 nm) in order to analyze the photoresponsive properties (Table 2). The molar extinction coefficients at λmax (εmax) of the π→π* transition bands show the order MDGA-AZ-MO < MDGA-AZ-F < MDGA-AZ-CA < MDGAAZ-NT < MDGA-AZ-CN. The molar extinction coefficient at 488 nm (ε488) also follows this order. On the other hand, for the molar extinction coefficients at 532 nm (ε532), MDGA-AZ-NT shows the highest ε value, which can be attributed to its absorption band appearing at the longer wavelength (λmax = 496 nm) compared with others. For the other azo molecular glasses, their ε532 values show the same order as that of their εmax values and the ε values at the wavelength of 589 nm are too small to be measured reliably.

|
Download:
|
| Fig. 2. The relationship between the absorbance and the concentration of MDGA-AZ-A (A: NT, CN, CA, F, MO) in DMF solutions obtained from the UV–vis spectra at four different wavelengths: (a) λmax, (b) 488 nm, (c) 532 nm, (d) 589 nm. | |
|
|
Table 2 Molar extinction coefficients ε (L·mol-1 cm-1) of MDGA-AZ-A (A: NT, CN, CA, F, MO), which were obtained from the experimental linear absorption-concentration relationship for the azo compounds in DMF solution. |

Upon exposure to the linearly polarized light or interference pattern formed by two laser beams, two types of photoresponsive variations are typically induced for the azo molecular glass films. Firstly, the polarized light irradiation will induce orientation of the azo chromophores through their repeated trans-cis-trans photoisomerization and optical birefringence of the films. Secondly, the SRG formation will be caused by the photoinduced mass transfer when the films are exposed to the light field with spatially varied intensity or polarization direction. These two types of photoresponsive behavior of the azo molecular glasses are discussed in the following separately.
Photoinduced birefringence of the azo molecular glasses was investigated using the experimental setup similar to that previously reported by Natansohn and Saishoji [41, 42]. Thin solid films of azo molecular glasses with smooth surfaces were prepared by spin-coating of their DMF solutions (30 wt%) to clean glass slides and vacuum drying at 60 ℃ for 48 h. The film thickness was controlled in a range from 800 to 900 nm by adjusting the spinning speed. The linearly polarized writing laser beam with the intensity of 50 mW/cm2 at three different wavelengths (λex = 488, 532 and 589 nm) was incident normal to the sample surfaces. The polarization direction of the laser beam was set at ± 45° with respect to the optical axes of two crossed polarizers in the optical path of the probe beam. The photoinduced change in the refractive index was probed in a real-time manner with a low-intensity beam from a He-Ne laser (632.8 nm). The angle between incident laser beam and probing laser beam was fixed to be 7°. The photoinduced birefringence was determined from the detected transmittance of the probe laser beam by using Eq. (1) [42],

|
(1) |
where I is the intensity of the transmitted probe laser beam when the sample film is irradiated with the writing beam, I0 is the intensity of the transmitted laser beam through the isotropic film without the polarizers, λ is the wavelength of the probe beam (632.8 nm), d is the film thickness (nm), and Δn is the birefringence.
Fig. 3 shows the birefringence variations when the films were irradiated with the linearly polarized laser beam at the three wavelengths λex = 488, 532 and 589 nm. A general tendency can be seen for the Δn variations of the molecular glass films in the writing process. The irradiation with the linearly polarized laser beam at 488 nm is discussed here as a typical example (Fig. 3a). Before the light irradiation, no transmission of the probing He-Ne laser beam (632.8 nm) is observed because the isotropic sample was placed between the crossed polarizers. When the writing laser beam is switched on (point A), the intensity of the transmitted laser beam suddenly increases in the first few seconds and then keeps gradual growth until saturation. It indicates that when starting the writing process, the optical anisotropy is quickly induced in the films and then increases gradually. Although the Δn variations show the similar tendency for the samples, as discussed below, the saturated values are significantly different for the azo molecular glasses. After switching off the writing beam (point B), the orientation relaxation can be observed and the relaxation degrees vary with the type of the azo molecular glasses. The Δn decay process is attributed to the relaxation of chromophores partially backing to the random orientation state due to the entropy increase tendency without the light irradiation. Finally, the stable states with obvious birefringence are achieved at point C.

|
Download:
|
| Fig. 3. Typical writing processes for the films of MDGA-AZ-A (A: NT, CN, CA, F, MO), irradiated with the incident laser beam at (a) 488 nm, (b) 532 nm, (c) 589 nm. The light intensity was 50 mW/cm2. | |
Some obvious distinctions can be seen for the different azo molecular glasses when irradiated at these three wavelengths. As shown in Fig. 3a, although the Δn increases can be seen for all the azo molecular glasses, the growth rates depend on the para-substituents on azobenzene units. After irradiation for several seconds, the Δn reaches saturation values of 0.080, 0.115, 0.110, 0.039 and 0.025 for MDGA-AZ-A (A: NT, CN, CA, F, MO) films, which shows the order MDGA-AZ-CN > MDGA-AZ-CA > MDGA-AZ-NT > MDGA-AZ-F > MDGA-AZ-MO. This order is consistent with those observed for other azo polymers and azo molecular glasses [39, 40, 43, 44], which shows that the photoinduced birefringence is mainly controlled by the azo chromophores. Fig. 3b shows the Δn variations of the azo molecular glass films when irradiated with the linearly polarized laser beam at 532 nm. MDGA-AZ-CN and MDGA-AZ-CA also show higher saturation Δn values of 0.056 and 0.049 compared with the others, but these values are obviously smaller than those obtained by irradiation with the 488 nm light. When irradiated with the linearly polarized laser beam at 589 nm (Fig. 3c), the photoinduced Δn values are extremely small and show trivial differences among the azo molecular glasses.
The birefringence growths can be fitted by a biexponential equation given below,

|
(2) |
where Δn is the birefringence achieved at time t, 1/ka and 1/kb are the characteristic times for birefringence growth, and A, B are two constants. The sum of A and B represents the maximum birefringence induced and the proportions of A and B to their sum (An and Bn) represent the relative contribution of each term to the induced birefringence. The fitting results for the birefringence growth are listed in Tables 3 and 4, where ka and kb correspond to the slow and fast growth processes, respectively. The fast and slow processes are attributed to the orientation of chromophores and motion of surrounding structures as a result of the repeated trans-cis-trans isomerization cycles [5, 41]. According to the parameters obtained from the curve fitting for the photoinduced birefringence kinetics at 488 nm (Table 3). The kb value of MDGA-AZ-CN is obviously larger than those of MDGA-AZ-NT, MDGA-AZ-CA, MDGA-AZ-F and MDGA-AZ-MO, and its ka is also the largest for the slow growth process. The combination of these factors results in the fast birefringence growth and high saturation value observed for MDGA-AZ-CN. Table 4 shows the curve fitting results of the photoinduced birefringence kinetics when irradiated with the light at 532 nm. The MDGA-AZ-CN also shows the largest kb value for the fast growth process, which corresponds to the fast birefringence growth. For the case of the irradiation with the 589 nm light, the photoinduced Δn values are extremely small (Fig. 3c), the data fitting cannot give reliable results.
|
|
Table 3 The parameters obtained from the curve fitting of the photoinduced birefringence kinetics of MDGA-AZ-A (A: NT, CN, CA, F, MO), obtained with the light irradiation at 488 nm. |

|
|
Table 4 The parameters obtained from the curve fitting of the photoinduced birefringence kinetics of MDGA-AZ-A (A: NT, CN, CA, F, MO), obtained with the light irradiation at 532 nm. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Above results show that MDGA-AZ-CN possesses the fastest birefringence growth rate and the high Δn when irradiated with the actinic light at 488 nm and 532 nm. Upon the irradiation at these two wavelengths, MDGA-AZ-CA also shows a reasonably good performance. The obviously higher efficiencies for these two materials compared with the others can be attributed to the more efficient trans-cis isomerization of the molecules when irradiated at these wavelengths [15, 43, 44]. It is believed that the irradiation wavelength of 488 nm corresponds to the overlapped n→π* and π→π* transition bands of the azo chromophores, which results in the efficient isomerization cycles for them. When the excitation wavelength increases from 488, 532 to 589 nm, the photoinduced birefringence of these two materials shows a gradual decline, which is attributed to the gradual decrease of the molar extinction coefficients and isomerization efficiency. The low birefringence values for MDGA-AZ-F and MDGA-AZ-MO can at least partially be attributed to the low light absorption efficiency at these three wavelengths. The only exception is the low Δn observed for MDGAAZ-NT, although it shows the strong light absorption in these wavelengths. Our previous study indicates that this exception can be reasonably explained by considering the strong dipole-dipole interaction between the azo chromophores bearing the nitro groups as the electron-withdrawing substituent [38].
The SRG formation behavior of azo molecular glasses was studied by using spin-coated films irradiated with interfering laser beams at three different wavelengths (488, 532 and 589 nm). SRG was inscribed by using the Lloyd mirror set-up according to the literatures [13, 14]. The linearly polarized laser beam, which was spatially filtered, expanded and collimated to have the intensity of 100 mW/cm2, was split by a mirror. One half of the p-polarized beam was reflected onto the film surface and coincident with the other half beam that directly struck the surface of the film to form an interference pattern. Fig. 4 shows typical AFM images (10 μm × 10 μm) of the SRGs inscribed on films of MDGA-AZ-A (A: NT, CN, CA), which were inscribed upon irradiation of interference patterns at the wavelengths of 488 nm and 532 nm for 1200s. It can be seen that the SRGs with the sinusoidal profile can be efficiently inscribed on the films, but the amplitudes of the gratings vary with the materials.

|
Download:
|
| Fig. 4. Typical AFM images (10 μm × 10 μm) of the surface patterns formed on (a) MDGA-AZ-NT, (b) MDGA-AZ-CN, (c) MDGA-AZ-CA films by the irradiation with 488 nm interfering laser beams; (d) MDGA-AZ-NT, (e) MDGA-AZ-CN, (f) MDGA-AZ-CA films by the irradiation with 532 nm interfering laser beams. The intensity of the light was 100 mW/cm2 and the irradiation time was 1200s. | |
{kind=link}
The first-order diffraction efficiency (DE) of the light-induced gratings was probed with a low power He-Ne laser (632.8 nm) in a real time mode and used to monitor the grating growth rate. The diffraction efficiency DE = Idiff /Iinc is defined as the ratio of the intensities of the first-order diffracted beam (Idiff) to the incident beam (Iinc) [13, 14, 45, 46]. Although the DE should also include the diffraction of the bulk phase grating, the diffraction of the SRG grating will be dominated when the amplitude is relatively large [47]. Fig. 5 shows typical diffraction efficiency as a function of time for the SRGs on the MDGA-AZ-A (A: NT, CN, CA, F, MO) films, irradiated with the interfering laser beams at 488, 532 and 589 nm. As shown in these figures, SRG formation is a relatively slower process compared with the photoinduced birefringence growth. The SRG formation behavior is also correlated to the substituents on azo chromophores and irradiation wavelengths, which are discussed below.

|
Download:
|
| Fig. 5. The first-order diffraction efficiency of the SRGs on the MDGA-AZ-A (A: NT, CN, CA, F, MO) films, irradiated with the interfering laser beams at (a) 488 nm, (b) 532 nm and (c) 589 nm. The light intensity was 100 mW/cm2. | |
{kind=link}
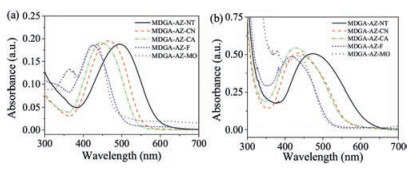
Among the azo molecular glasses, MDGA-AZ-CN with the cyano group as the electron-withdrawing substituent shows the fastest DE growth rate when irradiated with the interfering laser beams at 488 nm (Fig. 5a). It also shows the highest DE value (corresponding to the high grating amplitude) after irradiated for 1200s. MDGAAZ-CA also shows a reasonably high DE growth rate and high value when irradiated with the 488 nm light. Compared with those obtained by the irradiation at 488 nm, the SRG formation efficiency is obviously reduced when irradiated with the light at 532 nm, but MDGA-AZ-CN still shows the fastest DE growth rate compared with the others (Fig. 5b). On the other hand, the low growth rates and low DE values are observed for MDGA-AZ-F and MDGA-AZ-MO at these two irradiation wavelengths. Under the same light irradiation conditions, MDGA-AZ-NT shows the medium values of the DE growth corresponding to its SRG formation behavior. These results are consistent with the AFM observation as shown in Fig. 4. When the wavelength of the actinic light increases to 589 nm, no DE increase corresponding to the SRG formation can be seen for all the azo molecular glasses (Fig. 5c).
Previous investigations have indicated that the relative positions of the excitation wavelength (λex) and absorption bands show the significant effect on the SRGs growth rates [45, 46, 48]. Above results indicate that an efficient light absorption at λex is an important condition to show the highest grating formation rate. The fastest SRG formation rate can be achieved when the λex is longer than λmax, while there is still a significant absorption for the exciting light. This observation has also been attributed to the trans-cis isomerization efficiency, which depends on the competition between the excitation and relaxation [48]. It can be seen that MDGA-AZ-CN shows the highest SRG growth rate when irradiated at 488 nm. A similar tendency can also be observed for MDGA-AZCA. However, when λex increases towards the tail of the absorbance band, the SRG formation efficiency is obviously reduced. The low efficiency of MDGA-AZ-F and MDGA-AZ-MO for the SRG formation can at least be partially attributed to the low absorbance at λex. As there is almost no light absorption for the four azo molecular glasses MDGA-AZ-A (A: CN, CA, F, MO) at 589 nm, the irradiation cannot induce the SRG formation for these four azo molecular glasses. These observations further confirm that the trans-cis isomerization triggered by the light absorption is a necessary condition for the SRG formation. As the only exception, MDGA-AZ-NT shows the high light absorption intensity but the low efficiency for the SRG formation. Similar to the explanation given above for discussing the photoinduced birefringence result, it can be attributed to the strong dipole-dipole interaction between the azo chromophores [38]. An interesting result reported in a previous article is that the SRG can be enhanced upon annealing the nanoscale phase separation structure formed from a block copolymer of poly(ethylene oxide) and azobenzene-containing poly(methacrylate) at a higher temperature but below its Tg [49]. However, no such effect was observed for the current system due to the obvious difference in the molecular architecture.
Above results show that MDGA-AZ-CN possesses superior photoresponsive properties compared with others in the series. To demonstrate the feasibility of holographic data storage as a practical application, the holographic recording was tested by using the MDGA-AZ-CN film with the typical two-beam interference set-up. The optical set-up of the holographic recording is given in Fig. S7 in Supporting information. A diode-pumped frequency-doubled solid laser (532 nm) was used as the light source. The laser beam with Gaussian profile was split into two equal intensity beams by using a beam splitter to obtain the coherent object and reference waves. Both beams were linearly s-polarized (polarization perpendicular to the plane of incidence). A piece of the spin-coated film of MDGA-AZ-CN was exposed to the interference pattern formed by the coherent reference wave and object wave. One beam with its spot size controlled by a lens struck the recording media directly. The image of panda was carried by the other beam after being expanded by the two lenses. The object wave then struck the same area of the film after being collimated by the third lens. The whole holographic recording process was completed in 10–20 s. After recording the image into the MDGAAZ-CN film, a He-Ne laser (632.8 nm) beam was used as the reference wave to reconstructed the holographic pattern.
Fig. 6 gives the original picture and the holographic image reconstructed by the reference wave. It shows that the panda image is recorded as a hologram in the film. As shown above, the photoinduced volume birefringence and surface modulation have different time scales. In above process, the polarization modulation type of the holographic recording is exclusively achieved by controlling the irradiation time. In the recording process, the writing is completed in a short time to avoid possible surface modulation on the film. The image is recorded only by the photoinduced volume birefringence to accomplish the polarization holographic recording. Owing to the high Tg (96 ℃) of this recording material, the stability of the hologram is good.

|
Download:
|
| Fig. 6. (a) The original image of panda, (b) the holographic image read out from MDGA-AZ-CN film by the He-Ne laser (632.8 nm). | |
{kind=link}
In summary, a new class of azo molecular glasses with the fourarm architecture was prepared in this study. The azo molecules were synthesized to contain the different electron-withdrawing substituents on the azobenzene moieties in order to study the structure and property relationship. The absorption wavelength and the absorbance of the azo molecules are closely related to the electron-withdrawing substituents. The electron-withdrawing groups play a critically important role to affect the photoinduced birefringence and SRG formation properties of the azo molecular glasses. Among the azo molecular glasses, MDGA-AZ-CN and MDGA-AZ-CA show the highest birefringence growth rates and saturated values when irradiated with the linearly polarized light at 488 nm. Compared with the other four compounds, the azo molecular glass containing 4-cyanoazobenzene moieties (MDGAAZ-CN) shows the highest SRG formation rate and largest modulation amplitude when irradiated with the interfering laser beams. Especially, when irradiated with the actinic light at 488 nm, the SRG formation is observed to be very efficient. On the other hand, MDGA-AZ-F and MDGA-AZ-MO always show the low efficiency for photoinduced birefringence and SRG formation, which are not suitable to be used for such applications. Moreover, MDGA-AZ-CN shows the feasibility to be used as an optical datastorage medium, which is proved by the holographic recording.
AcknowledgmentThe financial support from the National Natural Science Foundation of China (Nos. 51773108 and 51233002) is gratefully acknowledged.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.01.015.
| [1] |
G.S. Kumar, D.C. Neckers, Chem. Rev. 89 (1989) 1915-1925. DOI:10.1021/cr00098a012 |
| [2] |
S. Xie, A. Natansohn, P. Rochon, Chem. Mater. 5 (1993) 403-411. DOI:10.1021/cm00028a003 |
| [3] |
J.A. Delaire, K. Nakatani, Chem. Rev. 100 (2000) 1817-1846. DOI:10.1021/cr980078m |
| [4] |
A. Natansohn, P. Rochon, Chem. Rev. 102 (2002) 4139-4175. DOI:10.1021/cr970155y |
| [5] |
N.K. Viswanathan, D.Y. Kim, S. Bian, et al., J. Mater. Chem. 9 (1999) 1941-1955. DOI:10.1039/a902424g |
| [6] |
T. Ikeda, J. Mamiya, Y. Yu, Angew. Chem. Int. Ed. 46 (2007) 506-528. DOI:10.1002/(ISSN)1521-3773 |
| [7] |
Y. Zhao, Macromolecules 45 (2012) 3647-3657. DOI:10.1021/ma300094t |
| [8] |
H. Yu, T. Ikeda, Adv. Mater. 23 (2011) 2149-2180. DOI:10.1002/adma.v23.19 |
| [9] |
S. Lee, H.S. Kang, J.K. Park, Adv. Mater. 24 (2012) 2069-2103. DOI:10.1002/adma.201104826 |
| [10] |
X.G. Wang, Azo Polymers: Synthesis, Functions and Applications, Springer, Berlin, 2017.
|
| [11] |
T. Ikeda, S. Horiuchi, D.B. Karanjit, S. Kurihara, S. Tazuke, Macromolecules 23 (1990) 42-48. DOI:10.1021/ma00203a009 |
| [12] |
T. Todorov, L. Nikolova, N. Tomova, Appl. Opt. 23 (1984) 4309-4312. DOI:10.1364/AO.23.004309 |
| [13] |
P. Rochon, E. Batalla, A. Natansohn, Appl. Phys. Lett. 66 (1995) 136-138. DOI:10.1063/1.113541 |
| [14] |
D.Y. Kim, S.K. Tripathy, L. Li, J. Kumar, App. Phys. Lett. 66 (1995) 1166-1168. DOI:10.1063/1.113845 |
| [15] |
H. Rau, Photoisomerization of azobenzenes, in: J.F. Rabek (Ed.), Photochemistry and Photophysics., vol. 2, CRC Press, Boca Raton, 1990, pp. 119-141.
|
| [16] |
P.C. Che, Y.N. He, X.G. Wang, Macromolecules 38 (2005) 8657-8663. DOI:10.1021/ma0511393 |
| [17] |
T.S. Lee, D.Y. Kim, X.L. Jiang, et al., Macromol. Chem. Phys. 198 (1997) 2279-2289. DOI:10.1002/macp.1997.021980720 |
| [18] |
M. Sukwattanasinitt, X. Wang, L. Li, et al., Chem. Mater. 10 (1998) 27-29. DOI:10.1021/cm970462k |
| [19] |
C.J. Barrett, A.L. Natansohn, P.L. Rochon, J. Phys. Chem. 100 (1996) 8836-8842. DOI:10.1021/jp953300p |
| [20] |
C.J. Barrett, P.L. Rochon, A.L. Natansohn, J. Chem. Phys. 109 (1998) 1505-1516. DOI:10.1063/1.476701 |
| [21] |
J. Kumar, L. Li, X.L. Jiang, et al., Appl. Phys. Lett. 72 (1998) 2096-2098. DOI:10.1063/1.121287 |
| [22] |
T.G. Pedersen, P.M. Johansen, N.C.R. Holme, P.S. Ramanujam, S. Hvilsted, Phys. Rev. Lett. 80 (1998) 89-92. DOI:10.1103/PhysRevLett.80.89 |
| [23] |
S. Bian, J.M. Williams, D.Y. Kim, et al., J. Appl. Phys. 86 (1999) 4498-4508. DOI:10.1063/1.371393 |
| [24] |
O.M. Tanchak, C.J. Barrett, Macromolecules 38 (2005) 10566-10570. DOI:10.1021/ma051564w |
| [25] |
K.G. Yager, C.J. Barrett, Macromolecules 39 (2006) 9320-9326. DOI:10.1021/ma061733s |
| [26] |
K.G. Yager, O.M. Tanchak, C. Godbout, H. Fritzsche, C.J. Barrett, Macromolecules 39 (2006) 9311-9319. DOI:10.1021/ma0617320 |
| [27] |
H. Nakano, T. Takahashi, T. Kadota, Y. Shirota, Adv. Mater. 14 (2002) 1157-1160. DOI:10.1002/1521-4095(20020816)14:16<1157::AID-ADMA1157>3.0.CO;2-Z |
| [28] |
Y. Shirota, J. Mater. Chem. 15 (2005) 75-93. DOI:10.1039/B413819H |
| [29] |
E. Ishow, C. Bellaïche, L. Bouteiller, K. Nakatani, J.A. Delaire, J. Am. Chem. Soc. 125 (2003) 15744-15745. DOI:10.1021/ja038207q |
| [30] |
M.J. Kim, E.M. Seo, D. Vak, D.Y. Kim, Chem. Mater. 15 (2003) 4021-4027. DOI:10.1021/cm034207d |
| [31] |
M.C. Guo, Z.D. Xu, X.G. Wang, Langmuir 24 (2008) 2740-2745. DOI:10.1021/la703091x |
| [32] |
L.M. Goldenberg, L. Kulikovsky, O. Kulikovska, J. Tomczyk, J. Stumpe, Langmuir 26 (2010) 2214-2217. DOI:10.1021/la9040562 |
| [33] |
Z.T. Nagy, B. Heinrich, D. Guillon, et al., J. Mater. Chem. 22 (2012) 18614-18622. DOI:10.1039/c2jm33751g |
| [34] |
J. Tomczyk, A. Sobolewska, Z.T. Nagy, et al., J. Mater. Chem. C 1 (2013) 924-932. DOI:10.1039/C2TC00627H |
| [35] |
K. Kreger, P. Wolfer, H. Audorff, et al., J. Am. Chem. Soc. 132 (2010) 509-516. DOI:10.1021/ja9091038 |
| [36] |
H. Audorff, R. Walker, L. Kador, H.W. Schmidt, Chem.-Eur. J. 17 (2011) 12722-12728. DOI:10.1002/chem.201100941 |
| [37] |
R. Walker, H. Audorff, L. Kador, H.W. Schmidt, Adv. Funct. Mater. 19 (2009) 2630-2638. DOI:10.1002/adfm.v19:16 |
| [38] |
J.J. Yin, G. Ye, X.G. Wang, J. Mater. Chem. C 1 (2013) 3794-3801. DOI:10.1039/c3tc30536h |
| [39] |
G. Ye, D.R. Wang, Y.N. He, X.G. Wang, J. Mater. Chem. 20 (2010) 10680-10687. DOI:10.1039/c0jm02228d |
| [40] |
Y.W. Wang, G. Ye, X.G. Wang, J. Mater. Chem. 22 (2012) 7614-7621. DOI:10.1039/c2jm00043a |
| [41] |
A. Natansohn, P. Rochon, J. Gosselin, S. Xie, Macromolecules 25 (1992) 2268-2273. DOI:10.1021/ma00034a031 |
| [42] |
A. Saishoji, D. Sato, A. Shishido, T. Ikeda, Langmuir 23 (2007) 320-326. DOI:10.1021/la061506j |
| [43] |
Y.W. Wang, Y.N. He, X.G. Wang, Polym. Bull. 68 (2012) 1731-1746. DOI:10.1007/s00289-011-0694-6 |
| [44] |
X.L. Wang, J.J. Yin, X.G. Wang, Polymer 52 (2011) 3344-3356. DOI:10.1016/j.polymer.2011.05.040 |
| [45] |
Y.N. He, X.G. Wang, Q.X. Zhou, Polymer 43 (2002) 7325-7333. DOI:10.1016/S0032-3861(02)00644-4 |
| [46] |
Y.Q. Zhou, B. Tang, X.G. Wang, Polymer 60 (2015) 292-301. DOI:10.1016/j.polymer.2015.01.033 |
| [47] |
A. Sobolewska, S. Bartkiewicz, A. Miniewicz, Schab-Balcerzak E., J. Phys. Chem. B 114 (2010) 9751-9760. DOI:10.1021/jp103756h |
| [48] |
M.J. Kim, J. Lee, C. Chun, et al., Macromol. Chem. Phys. 208 (2007) 1753-1763. DOI:10.1002/(ISSN)1521-3935 |
| [49] |
H.F. Yu, K. Okano, A. Shishido, et al., Adv. Mater. 17 (2005) 2184-2188. DOI:10.1002/(ISSN)1521-4095 |