 2019, Vol. 30
2019, Vol. 30 


b College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China;
c State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China;
d Department of Nuclear Medicine, Jinling Hospital, School of Medicine, Nanjing University, Nanjing 210002, China
Mesoporous materials, especially periodic mesoporous organosilica (PMO) nanoparticles, have attracted increasing attention in biomedical fields because of their colorful frameworks, high surface area, large pore volume, uniform pore size, and excellent biocompatibility [1-17]. Once exposing to physiological environments, the negatively charged PMOs tend to interact with ions and plasma proteins, resulting in severe agglomeration and increase of particle diameter. Thus, the PMOs are more easily recognized and cleared by biological immune system, resulting in a significant reduction in treatment efficacy for diseases [18-21].
Modification of PMOs with hydrophilic and biologically compatible polymers can reduce the adsorption for plasma proteins, prolong the blood circulation, and increase targeting ability for diseases [22-27]. For example, polyethylene glycol (PEG) modified PMOs show significantly improved in vivo biocompatibility [28]. Modification of polyetherimide and cell-penetrating peptide transactivator of transcription (TAT) endows PMOs with enhanced transfection efficiencies for plasmids and intranuclear gene transport capability [29]. Recently, Lu et al. report that the PMOs conjugated with PEG and anti-Her2 affibody shows excellent targeting property for breast cancer cells [30]. However, few of these studies succeed in showing the stability of PMOs in physiological environments. In addition, although PEG has been widely used to modify PMOs for biomedical applications [31-33], the influence of molecular weight of PEG on the stability of PMOs has not been reported to the best of our knowledge.
Herein, thioether-bridged periodic mesoporous organosilica nanopspheres are synthesized by a cetyltrimethylammonium bromide (CTAB)-directing sol–gel process. Then the PMOs are modified with different molecular weighted PEG via click reaction for the first time. The successful modification of the PEG on the PMOs is confirmed by Fourier transform infrared spectroscopy (FIIR) and thermogravimetric analysis. Furthermore, the effects of PEG molecular weight on the stability of the PEGylated PMOs in phosphate buffer (PBS) and Dulbecco's modified Eagle's medium (DMEM) are investigated using dynamic light scattering measurements. The chain length of the PEG great affects the stability of PMOs in PBS solutions, and the PMO-PEG2K has best stability and dispersion in both PBS and DMEM.
Cetyltrimethylammonium bromide (CTAB, ≥ 99%), concentrated ammonia aqueous solution (25–28wt%), anhydrous ethanol, triphenylphosphine (≥ 99%), tetraethoxysilane (TEOS, ≥ 28.4%), and concentrated hydrochloric acid (37%) were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Bis[3-(triethoxysilyl)propyl] tetrasulfide (TESPTS, ≥ 90%) was obtained from Sigma–Aldrich (St. Louis, MO, USA). Deionized water (Millipore) with a resistivity of 18 MV·cm was used in all experiments. Methoxy polyethylene glycol-maleimide (mPEGMAL, ≥ 90%) with different molecular weights was purchased from Shanghai Seebio Biotech, Inc. (China).
Thioether-bridged PMOs were prepared via a CTAB-directing sol -gel process. In brief, 0.16 g of CTAB was dissolved in a mixture containing of ethanol (30 mL), water (100 mL), and concentrated ammonia aqueous solution (1 mL, 25wt%) and stirred at 35 ℃. Afterward, mixed precursors of TEOS (0.25 mL) and TESPTS (0.25 mL) were added. After reaction for 24 h, the products were collected by centrifugation and washed with ethanol three times. The products were extracted in a solution containing 100 mL of ethanol and 200μL of concentrated HCl at 60 ℃ for 3 h to remove the CTAB surfactant.
mPEG-MAL was connected to the surface of the thioether-bridged PMOs by click reaction. In brief, 0.065 g of PMOs was dispersed in a mixed solution containing 1.1 mL of dioxane and 0.3 mL of water. The mixture was then added with 0.1 g of triphenylphosphine and heated to 40 ℃. Afterward, 40μL of concentrated hydrochloric acid was added under nitrogen and the reaction was proceeded for 2 h. The product was washed with ethanol three times and stored in 2 mL of ethanol. mPEG-MAL dissolved in 12 mL of water was added to the obtained PMOs dispersed in 2 mL of ethanol. After shaking overnight reaction and washing with water three times, the product PMO-PEG was obtained. The concentrations for the mPEG-MAL with PEG350, PEG500, PEG1k, PEG2k, PEG5k and PEG10k are 0.047, 0.068, 0.14, 0.27, 0.68, and 1.35 mg/mL, respectively. The products were abbreviated as PMO-PEG350, PMO-PEG500, PMO-PEG1k, PMO-PEG2K, PMO-PEG5k, and PMO-PEG10k, respectively.
The graft density of PEG was calculated according to formulas 1-3.
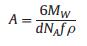
|
(1) |
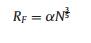
|
(2) |
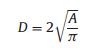
|
(3) |
where A is the area occupied by single graft PEG, nm-2; d is the distance between two adjacent PEG sites, nm; MW is the molecular weight of PEG; NA is Avogadro's constant; f is the mass fraction of PEG; ρ is the density of nanoparticles, RF is the Flory radius of PEG; α isthe length of thePEG monomer, α =0.35 nm; N isthe numberof PEG monomer; D is the diameter of the nanoparticles.
The stability of PMO-PEG was investigated in PBS and DMEM. The unmodified PMOs, PMO-PEG350, PMO-PEG500, PMO-PEG1k, PMO-PEG2k, PMO-PEG5k, and PMO-PEG10k were dispersed in PBS or DMEM, respectively. The hydrodynamic diameters and polydispersity index (PDI) of the nanoparticles were measured and recorded at different time points at 37 ℃.
Transmission electron microscopy (TEM) images were obtained on an HT7700 microscope (Hitachi, Tokyo, Japan) at 100kV. For TEM observation, an ethanol solution of the thioether-bridged PMOs was dripped on carbon coated copper mesh and dried at room temperature. The nitrogen adsorption and desorption isotherms of the PMOs were measured at -196 ℃ using a Micromeritics ASAP 2020 analyzer. The samples were degassed in a vacuum at 150 ℃ for 12 h before the measurements. The specific surface area PMOs were calculated by Brunauer-EmmettTeller (BET) method. The pore size distributions were derived from the adsorption branches of isotherms by applying proper BarrettJoyner-Halenda (BJH) method. Fourier transform infrared (FT-IR) spectra were measured on a Nicolet NEXUS870 spectrometer (Nicolet Instruments Inc. Madison, WI, USA) using KBr pellets of the solid samples. For FT-IR measurements, a small amount of pure potassium bromide was mixed with samples, ground evenly, and pressed to make pellets. 29Si magic-angle spinning (MAS) NMR spectra were recorded at 9.47T using a Bruker AVIII400 spectrometer operating with a frequency of 79.48MHz, a spin rate of 6.0kHz, and a recycle delay of 120s. Zeta potential and the hydrodynamic diameters of the samples were measured on a Brookhaven ZetaPALS analyzer.
Thioether-bridged PMO nanospheres are synthesized via CTAB-directing sol-gel process using TESPTS and TEOS as coprecursors. Low-magnification TEM images show that the thioether-bridged PMO nanospheres have a uniform spherical shape with a diameter of approximately 61 nm (Figs. 1a and b). High-magnification TEM image shows the thioether-bridged PMOs is porous in structure (Fig. 1c). The composition and mesostructured of the PMO nanospheres were further characterized by FT-IR, NMR, and nitrogen adsorption. The FT-IR spectrum of the thioether-bridged PMO nanospheres shows the characteristic band of Si—O—Si at 1068 cm-1 and the band of disulfide bond (-S-S-) at 576 cm-1, suggesting the thioether-bridged organosilica frameworks (Fig. 2a). 29Si NMR spectrum of the thioether-bridged PMOs shows a setof peaksat -47, -56, and -64 ppm, corresponding to T1 (C—Si (OSi)1(OX)2 X=H or Et), T2 (C-Si (OSi)2(OX) and T3 (C—Si (OSi)3), respectively (Fig. 2b). The peaks at -88, -99, -107 ppm correspond to Q2 (Si(OSi)2(OX)2), Q3 (Si(OSi)3(OX)) and Q4 (Si(OSi)4)), respectively. The silicon atoms located at T and Q are 51.8% and 48.2%, respectively. The nitrogen isotherms of the thioether-bridged PMO nanospheres show a type Ⅳ curve with a large hysteresis loop, suggesting typical mesostructure (Fig. 3c). The specific surface area and pore size of the PMO nanospheres are calculated to be 217 m2/g and 3.5 nm (Fig. 3d), respectively.

|
Download:
|
| Fig. 1. TEM images of the thioether-bridged PMO nanospheres synthesized via CTAB-directing sol -gel process. | |

|
Download:
|
| Fig. 2. (a) FT-IR spectra, (b) 29Si NMR spectra, (c) nitrogen sorption isotherms and (d) pore size distribution curves of the thioether-bridged PMO nanospheres. | |

|
Download:
|
| Fig. 3. (a) FT-IR spectra and (b) thermogravimetric analysis of the PMOs and PMOPEG. | |
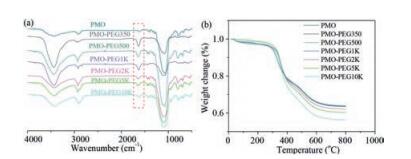
The modification of different molecular weighted PEG on the PMO was confirmed by FT-IR and thermogravimetric analysis. The FT-IR spectra show that the PMO-PEG350, PMO-PEG500, PMO-PEG1k, PMO-PEG2k, PMO-PEG5k, and PMO-PEG10k haverelatively strong peaks at 1635 cm-1, which is assigned to the tertiary amide bond in maleimide. The FT-IR results demonstrate that the PMOs have been successfully modified with PEG by click reaction. The thermogravimetric analysis shows that the PMOs and PMO-PEG have a weight loss at 300–400 ℃, which is attributed the thermolysis of disulfide bonds in the frameworks. The weight loss at 400–600 ℃ is assigned to the breakage of the C—C bonds and PEG chain.
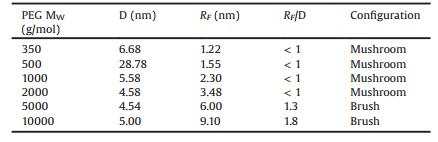
The parameters of the PEG modified on the PMO nanospheres are calculated and shown in Table 1. When the molecular weight is 350–2000, the PEG on the surfaces of the PMOs forms mushroom structures. When the molecular weight is up to 5000–10000, the PEG forms brush structures. The PEG modification on the hydrodynamic diameter of the PMOs was further studied. The hydrodynamic diameter of the PMOs measured by DLS is 168 nm (Fig. 4a), which is larger than that observed by TEM. On one hand, the larger diameter measured by DLS is due to the presence of hydration layer in aqueous solution. On the other hand, the thioether-bridged PMO nanospheres contain high content of organic moieties, and thus they have relatively low surface potentials, resulting in closing each other and even agglomerating in aqueous solution. Thus, the particle diameter of the PMOs is larger than the TEM results. After modification PEG, the hydrodynamic diameters slightly increase because PEG have a longer chain length, which increases hydration layers. As the molecular weight of PEG increases, larger particle diameters for the PMO-PEG are observed. The zeta potential of the thioether-bridged PMOs is measured to be -13.76 mV (Fig. 4b), which is low than that of the classical mesoporous silica due to the decrease of Si—OH groups on the particle surface. With the increase of the molecular weight of PEG, the absolute value of zeta potentials for the PMO-PEG slightly increases.
|
|
Table 1 Parameters of PEG decorated on the PMO nanospheres. |
{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Fig. 4. Hydrodynamic diameters and zeta potentials of PMOs and PMO-PEGs. | |
{kind=link}
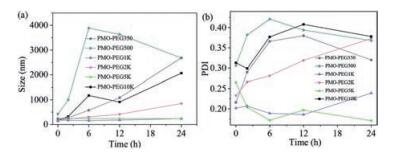
We tested the stability of the PMO-PEGs with different PEG molecular weights in PBS. The hydrodynamic diameters of the PMO-PEG350 and PMO-PEG500 increase significantly after 6 h in PBS, suggesting they aggregate in PBS solution with high ion concentration (Fig. 5). This is because the modified PEG on the PMO-PEG350 and PMO-PEG500 forms a mushroom-shaped configuration. In addition, the short PEG chain on the surface cannot provide enough steric hindrance. Notably, PMO-PEG1K, PMO-PEG2K and PMO-PEG5K have better stability in PBS solution, indicating that the increase of chain length of PEG can effectively reduce the interaction between particles and ions, prolonging the stability and dispersion. In addition, PMO-PEG10k shows relatively low stability, suggesting appropriate chain length for the modified PEG is important for improving their solution stability.

|
Download:
|
| Fig. 5. The changes of hydrodynamic diameters and PDI of the PMO-PEGs with time in PBS. | |
{kind=link}
We further investigated the influence of molecular weight of PEG on the stability of the PMO-PEGs in DMEM. The dynamic light scattering test shows that the size distribution of the PMO-PEG350 and PMO-PEG500 in DMEM is fluctuating with time (Fig. 6). The results are attributed to that the PMO-PEG350 and PMO-PEG500 adsorb the nutrients such as amino acids and protein in DMEM, which decreases the stability. The diameters of the PMO-PEG5K and PMO-PEG10 K also increase at 2 h, suggesting extremely long PEG chain cannot improve the stability of PMO nanospheres in solution. Notably, the PMO-PEG1K are stable in DMEM for 12 h. Furthermore, the PMO-PEG2k keep stable up to 24 h in DMEM, suggesting the stability can be improved by adjusting the PEG chain length.

|
Download:
|
| Fig. 6. The changes of hydrodynamic diameters and PDI of the PMO-PEGs with time in DMEM. | |
{kind=link}
In summary, we have synthesized thioether-bridged periodic mesoporous organosilica (PMO) nanospheres via a CTAB-directing sol - gel process. The PMOs have been further modified with different molecular weights of PEG via click reactions for the first time. The successful connection of the PEG on the PMOs was confirmed by FT-IR spectra and thermogravimetric analysis. The hydrodynamic diameter and PDI of the PMO-PEG with different molecular weights of PEG in PBS and DMED were measured. The results demonstrate that the PMO-PEG1K, PMO-PEG2K and PMO-PEG5K have better stability in PBS solution. The PMO-PEG2K has the best stability and dispersity in DMEM. Taken together, this work demonstrates a facile and effective PEGylation method for PMO nanoparticles. Furthermore, the stability investigations for the different molecular weighted PEG modified PMO provide important knowledge to guide the modification of PMO for biomedical applications.
AcknowledgmentsWe greatly appreciate the financial support from the Natural Science Foundation of Jiangsu Province (Nos. BK20160017 and BK20160610), the National Natural Science Foundation of China (Nos. 21603106, 51822202 and 51772050), the State Key Laboratory of Analytical Chemistry for Life Science (No. 5431ZZXM1717), Shanghai Rising-Star Program (No. 18QA1400100), Youth Topnotch Talent Support Program of Shanghai, DHU Distinguished Young Professor Program and Fundamental Research Funds for the Central Universities.
| [1] |
Z.G. Teng, X.D. Su, Y. Zheng, et al., Chem. Mater. 25 (2013) 98-105. DOI:10.1021/cm303338v |
| [2] |
Z.G. Teng, X.D. Su, B.H. Lee, et al., Chem. Mater. 26 (2014) 5980-5987. DOI:10.1021/cm502777e |
| [3] |
Z.G. Teng, J.J. Zhang, W. Li, et al., Small 12 (2016) 3550-3558. DOI:10.1002/smll.201600616 |
| [4] |
J. Tao, M. Dang, X.D. Su, et al., J. Colloid. Interf. Sci. 527 (2018) 33-39. DOI:10.1016/j.jcis.2018.05.024 |
| [5] |
Y. Chen, J.L. Shi, Adv. Mater. 28 (2016) 3235-3272. DOI:10.1002/adma.201505147 |
| [6] |
F. Hoffmann, M. Cornelius, J. Morell, M. Froba, Angew. Chem. Int. Ed. 45 (2006) 3216-3251. |
| [7] |
J.G. Croissant, X. Cattoën, M.W.C. Man, et al., Nanoscale 7 (2015) 20318-20334. DOI:10.1039/C5NR05649G |
| [8] |
J.G. Croissant, Y. Fatieiev, H. Omar, et al., Chem. Eur. J. 22 (2016) 1-10. DOI:10.1002/chem.201504553 |
| [9] |
X.R. Zhou, X.W. Cheng, Y.H. Zhu, et al., Chin. Chem. Lett. 29 (2018) 405-416. DOI:10.1016/j.cclet.2017.06.021 |
| [10] |
L. Yu, W.L. Liu, Z.F. Zhang, Z.T. Song, Chin. Chem. Lett. 26 (2015) 700-704. DOI:10.1016/j.cclet.2015.01.039 |
| [11] |
P. Han, T.C. Liu, X.W. Ji, S.K. Tang, Chin. Chem. Lett. 29 (2018) 1305-1309. DOI:10.1016/j.cclet.2017.10.042 |
| [12] |
J.T. Dai, Y. Zhang, H.C. Lia, et al., Chin. Chem. Lett. 28 (2017) 531-536. DOI:10.1016/j.cclet.2016.11.008 |
| [13] |
J. Wei, Y. Ren, W. Luo, et al., Chem. Mater. 29 (2017) 2211-2217. DOI:10.1021/acs.chemmater.6b05032 |
| [14] |
J. Wei, Z.K. Sun, W. Luo, et al., J. Am. Chem. Soc. 139 (2017) 1706-1713. DOI:10.1021/jacs.6b11411 |
| [15] |
G. Wang, J. Qin, X.R. Zhou, et al., Adv. Funct. Mater. 28 (2018) 1806144. DOI:10.1002/adfm.v28.51 |
| [16] |
Y. Rui, X.M. Wu, B.D. Ma, et al., Chin. Chem. Lett. 29 (2018) 1387-1390. DOI:10.1016/j.cclet.2017.10.033 |
| [17] |
P. Han, T.C. Liu, X.W. Ji, et al., Chin. Chem. Lett. 29 (2018) 1305-1309. DOI:10.1016/j.cclet.2017.10.042 |
| [18] |
A. Salvati, A.S. Pitek, M.P. Monopoli, et al., Nat. Nanotechnol. 8 (2013) 137-143. DOI:10.1038/nnano.2012.237 |
| [19] |
A.E. Nel, L. Mädler, D. Velegol, et al., Nat. Mater. 8 (2009) 543-557. DOI:10.1038/nmat2442 |
| [20] |
T. Cedervall, I. Lynch, S. Lindman, et al., Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 2050-2055. DOI:10.1073/pnas.0608582104 |
| [21] |
X. Du, F. Kleitz, X.Y. Li, Adv. Funct. Mater. 28 (2018) 1707325. DOI:10.1002/adfm.201707325 |
| [22] |
R. Gref, M. Luck, P. Quellec, et al., Colloids Surf. B Biointerfaces 18 (2000) 301-313. DOI:10.1016/S0927-7765(99)00156-3 |
| [23] |
P. Aggarwal, J.B. Hall, C.B. McLeland, et al., Adv. Drug Deliver. Rev. 61 (2009) 428-437. DOI:10.1016/j.addr.2009.03.009 |
| [24] |
T.A. Horbett, Cardiovasc. Pathol. 2 (1993) 137-148. |
| [25] |
G. Prencipe, S.M. Tabakman, K. Welsher, et al., J. Am. Chem. Soc. 131 (2009) 4783-4787. DOI:10.1021/ja809086q |
| [26] |
N. Mizoshita, T. Tani, S. Inagaki, Chem. Soc. Rev. 40 (2011) 789-800. DOI:10.1039/C0CS00010H |
| [27] |
X. Duan, Y.P. Li, Small 9 (2013) 1521-1532. DOI:10.1002/smll.201201390 |
| [28] |
P. Huang, Y. Chen, H. Lin, et al., Biomaterials 125 (2017) 23-37. DOI:10.1016/j.biomaterials.2017.02.018 |
| [29] |
M.Y. Wu, Q.S. Meng, Y. Chen, et al., Adv. Mater. 27 (2015) 215-222. DOI:10.1002/adma.v27.2 |
| [30] |
N. Lu, Y. Tian, W. Tian, et al., ACS Appl. Mater. Interfaces 8 (2016) 2985-2993. DOI:10.1021/acsami.5b09585 |
| [31] |
H. Otsuka, Y. Nagasaki, K. Kataoka, Adv. Drug Deliver. Rev. 64 (2012) 246-255. DOI:10.1016/j.addr.2012.09.022 |
| [32] |
Z.G. Teng, C.Y. Wang, Y.X. Tang, et al., J. Am. Chem. Soc. 140 (2018) 1385-1393. DOI:10.1021/jacs.7b10694 |
| [33] |
G.S. Irmukhametova, B.J. Fraser, J.L. Keddie, et al., Langmuir 28 (2012) 299-306. DOI:10.1021/la2038735 |