 2019, Vol. 30
2019, Vol. 30 


Peptides as a class of molecules lying between proteins and small molecules in size, and integrating the advantages of both, are increasingly being explored as potential drugs for targeted cancer therapy [1-5]. Although promising, the use of peptides for therapeutic modulation of intracellular targets can be strongly hampered by their lack of resistance to proteolytic degradation and the low efficiency of cellular uptake [6-8]. However, strategies to improve the proteolytic stability of peptides have been rapidly developed, including peptide cyclization, backbone/side chain modification, and unnatural residue substitution [9-19]. Among these, cyclization to create constrained peptides with a rigid secondary or tertiary structure (e.g., α-helix) is particularly attractive [20-24], because it not only drastically improves the proteolytic stability of peptides, but can create peptides with structures globally complementary in shape to the binding domain of targets. The efficiency of cellular uptake of peptides can also be increased through modulation of peptide charges, secondary structures, and hydrophobicity [25-27]. In this context, constrained peptide templates that are tolerant to extensive sequence manipulation and amenable to bioactive peptide design would be of great value to the development of new peptide therapeutics and ligands.
The α-helix is the most abundant secondary structural motif in proteins, and is involved in many protein-protein interactions [6, 28-30]. The α-helical structure of a bioactive sequence can usually be efficiently stabilized through cyclization with an organic linker, so as to improve its ability to target the specific protein [31-35]. Of the many α-helical peptides with therapeutic potentials, those developed to inhibit the p53-MDM2 interaction are particularly potent, and at least one of them has been progressed to clinical trials [12, 20, 36, 37]. p53 is one of the most important tumor suppressor, but its function is inactivated in virtually all cancers [38-40]. In ~50% of cancers, the inactivationof p53 is caused by the overexpression of MDM2 and/or its homolog MDMX [37]. Interestingly, peptide inhibitors that are developed for MDM2 can usually target both proteins [20, 37]. To develop potent peptide inhibitors for MDM2, as an intracellular target, a stable α-helical scaffold that is not only highly resistant to proteolysis, but amenable to extensive manipulation of sequence for optimization of activity and cellular uptake efficiency are greatly desired.
Recently, we have reported that the proteolytic (and structural) stability of peptides can be significantly enhanced by controllably dimerizing them into α-helical dimers with two disulfide bonds [41]. However, disulfide bonds are not stable under the intracellular reducing conditions [42-49], thus disabling the dimeric α-helices as structurally stable scaffolds for the design of inhibitors towards intracellular targets. In this study, by replacing disulfide bonds with physiologically stable chemical bonds (i.e., bisthioether crosslinkers), we create a novel interchain doubly-bridged α-helical scaffold (Fig. 1), which allows us to develop potent inhibitors for the p53-MDM2 interaction with extremely high proteolytic stability. As dimeric α-helices consist of two identical bioactive sequences, and one of the two only exerts, in principle, an efficacy of stabilizing the α-helical structure, but not directly interacts with the targets, its sequence can thus be extensively manipulated to improve the efficiency of cellular uptake, without sacrificing the binding capability of the α-helical dimer.

|
Download:
|
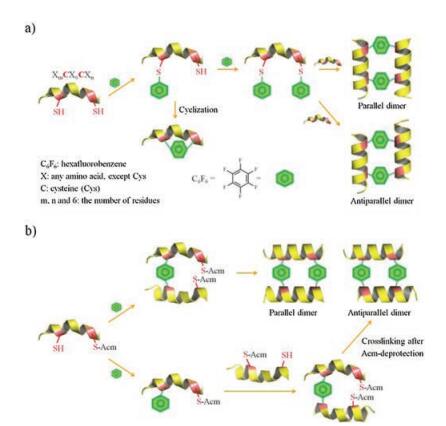
| Fig. 1. Two different strategies to design and synthesize interchain doubly-bridged α-helical peptide dimers. a) This strategy leads to indistinguishable formation of both antiparallel and parallel dimers and suffers from the competitive intrachain cyclization. b) This strategy benefits from the orthogonal Acm-protection to avoid the problem of intrachain cyclization, and enables the selective formation of antiparallel or parallel dimers. | |
{kind=link}
Secondary structural motifs can be stabilized by cyclizing peptides with organic crosslinkers. To stabilize a given bioactive epitope, the length of organic crosslinkers has to match the structural gap of peptides. This has been found challenging, which may require extensive optimization of the crosslinker length and the position for the crosslinker placement. Alternatively, peptide secondary structures can be stabilized by a covalent dimerization of peptides with interchain crosslinking, involving the mutual locking of the pre-organized structure of the two symmetric peptide chains. One example of particular interest is the dimeric α-helices stabilized by two interchain disulfide bonds [41, 45, 50]. The construction of these dimeric α-helices relies on the oxidationinduced dimeric folding of peptides with two substituted i and i+7 cysteines, which involve no optimization of the crosslinker length and position. To replace the reduction-sensitive cystine bridges with stable bisthioether crosslinkers, there are two facile strategies that can be explored. The first strategy relies on the modification of the i and i+7 cysteines-bearing peptide with two bifunctional crosslinkers first, and the subsequent crosslinking reaction of this modified peptide with an unmodified peptide (Fig. 1a). The second strategy relies on the crosslinking of two biscysteine-bearing peptides (one of the two cysteines orthogonally protected by acetamidomethyl (S-Acm)) with a crosslinker first, and the subsequent introduction of the second crosslinker after Acm deprotection (Fig. 1b). Both strategies have their own merits and demerits. The first strategy can afford the final products with fewer reaction steps, which however involves the competitive sidereaction formation of monocyclic peptides during the first step of bis-crosslinker modifications. In addition, this strategy yields both parallel and antiparallel dimers, though the one with more favorable interchain non-covalent interactions can be expected to be the major product. The second strategy is more sophisticated, but it enables the synthesis of the desired parallel or antiparallel dimer as a single product, and it allows us to incorporate two different crosslinkers into the dimers.
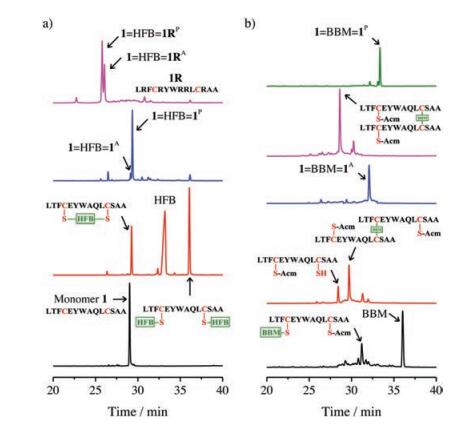
We chose several different crosslinkers reported in literatures [21, 33, 36], including 4, 4'-bis(bromomethyl)diphenyl, 1, 3-bis(bromomethyl)benzene, 1, 4-bis(bromomethyl)benzene, 1, 2-bismaleimidoethane, 1, 3-butadiene diepoxide and hexafluorobenzene (HFB), to test the feasibility of the first strategy to synthesize peptide dimers. A peptide (1; sequence: LTFCEYWAQLCSAA) has the potential to inhibit the p53-MDM2 interaction was used as a model peptide [20, 41]. This peptide can bind to a large cleft present at the surface of MDM2 and adopt an α-helical conformation. Three residues in this peptide, F3, W7 and L10, were important to the protein binding [51], which locate on a single face of an α-helix. Thus, the introduction of two i and i+7 cysteines on the faces distant from the binding interface does not affect the protein binding capability. We found that reaction of biscysteine-bearing peptides with bis-bromomethyl compounds only results in the formation of intra-chain cyclized products (data not shown), suggesting that the intermolecular modification failed to compete with the intramolecular cyclization to afford the doubly-modified peptide for subsequent dimerization. Though 1, 3-butadiene diepoxide can afford the doubly-modified peptides, the peptide is not stable for HPLC purification, and we can only obtain purified peptide with dual HFB modification (Fig. 2a). Thus, the HFB-modified peptide was then used to synthesize the dimeric peptide by reacting with the unmodified biscysteine-bearing peptide, and we observed the formation of a parallel dimer (1=HFB=1P) as the major product (Fig. 2a and Supporting information). Compared to the bis-bromomethyl crosslinkers, HFB modified onto the peptides should be more rigid in structure, which might limit the intra-chain cyclization, and thus facilitating the further modification of the peptides with the second HFB. Heterodimers can be synthesized by the dimerization of the HFBmodified peptide with another biscysteine-bearing peptide (e.g., peptide 1R; sequence: LRFCRYWRRLCRAA). 1R is derived from the sequence of peptide 1 by substituting residues distant from the peptide-MDM2 binding interface with arginines. As shown in Fig. 2a and Fig. S3 (Supporting information), both parallel and antiparallel dimers (1=HFB=1RP and 1=HFB=1RA) were obtained after the dimerization without an isomer deviation as obvious as that observed from the previous homodimer. The second strategy for dimeric peptide synthesis is more tolerant to the type of crosslinkers. By using this strategy, we can synthesize dimeric peptides crosslinked with 1, 4-bis(bromomethyl)benzene crosslinker (BBM). Based on HPLC chromatograms shown in Fig. 2b, both parallel and antiparallel dimers (1=BBM=1P and 1=BBM=1A, respectively, Fig. S5 in Supporting information) can be obtained with good to excellent yields. In addition, both 1=HFB=1P and 1=HFB=1A can be synthesized using this strategy (Fig. S6 in Suopporting information, enabling us to compare the structure and property of these two dimers.

|
Download:
|
| Fig. 2. (a) HPLC chromatograms showing the synthesis of the dimeric peptides 1=HFB=1P, 1=HFB=1RP and 1=HFB=1RA using the first strategy shown in Fig. 1. (b) HPLC chromatograms showing the synthesis of the dimeric peptides 1=BBM=1P and 1=BBM=1A using the second strategy shown in Fig. 1. All intermediates and products were characterized using mass spectrometry. The experimental details were given in the experimental section of the Supporting information. | |
{kind=link}
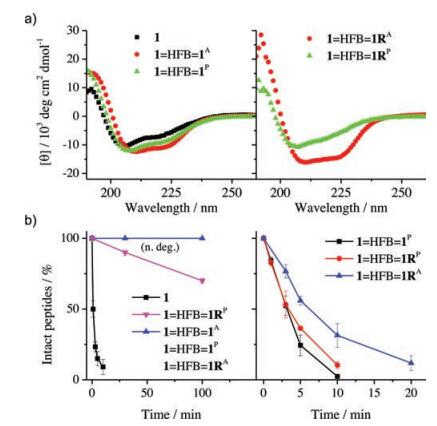
To examine if the dimerization of peptides by organic crosslinkers could stabilize the peptides into the α-helical structure, the peptide dimers was isolated and analyzed by CD spectroscopy. As seen in Fig. 3a, CD spectra of the peptide dimers display typical characteristics of α-helical secondary structure, which consist of strong negative (at 208 and 222 nm) and positive (195 nm) bands. Moreover, in all cases the antiparallel dimers have a slightly higher α-helicity compared to the corresponding parallel dimers. Resistance to proteolysis of the dimeric α-helical peptides was then evaluated and compared by monitoring the kinetics of degradation by HPLC. Kinetic curves of degradation were plotted as the percentage of intact peptide measured by analysis of peak areas as a function of time (Fig. 3b). Amino acid residues F, Y and W in 1 were the cleavage sites for chymotrypsin. We found that monomeric 1 can be rapidly cleaved under a lower concentration of chymotrypsin (0.1 μmol/L), but the dimers kept intact or were cleaved very slowly. The dimers can only be rapidly cleaved under higher concentrations of chymotrypsin (e.g., 10 μmol/L, Fig. 3b). These results clearly show that the interchain double bisthioether bridges can indeed lock linear peptides into α-helical structures, and significantly increase the proteolytic stability of peptides. These interchain doubly-bridged α-helical peptides provide an interesting platform for the development of potent protein binders with high structural and proteolytic stability.

|
Download:
|
| Fig. 3. a) CD spectra of 1 (dissolved in phosphate buffer with 5% v/v acetonitrile; pH 7.4) and peptide dimers (dissolved in phosphate buffer, pH 7.4); peptide concentration is 30 μmol/L. b) Kinetics of peptide degradation in the presence of 0.1 (left) and 10 (right) mmol/L chymotrypsin in phosphate buffer (pH 7.4); peptide concentration is 50 μmol/L; data were presented as mean ± s.d. (n = 3). n. deg.: no degradation of dimeric peptides except 1=HFB=1RP. | |
{kind=link}
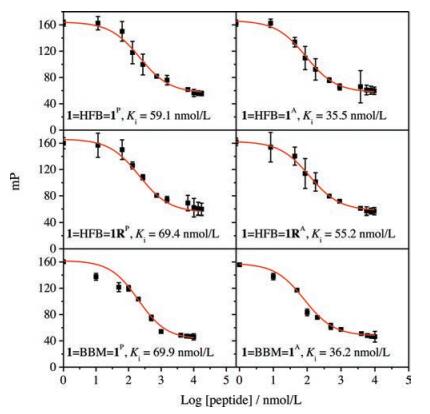
We then examined if the interchain doubly-bridged α-helical peptides could retain the binding affinity towards MDM2 by using a well-established fluorescence polarization competition assay (Fig. 4) [41, 52]. Interestingly, all dimers exhibit a comparable or slightly higher affinity to MDM2 (Ki < 100 nmol/L, Fig. 4) compared to the monomer (Ki = 113 ± 58 nmol/L) [41], likely due to the enhanced α-helicity of these dimeric peptides. To our surprise, even the heterodimers (1=HFB=1RP and 1=HFB=1RA) display a high binding affinity to MDM2 as well (Fig. 4), though one of their peptide chains have been extensively mutated, suggesting very likely that the presence of one MDM2-binding peptide chain in dimeric α-helices is sufficient for retaining the binding affinity. This is interesting for the design of protein binders for applications in biological environments, because the tunability of the sequence of one of the dimeric peptide chains would enable the further optimization of not only the affinity and stability of the interchain doubly-bridged α-helical peptides, but also the cellular uptake efficiency of these peptides when applications in the intracellular environments are desired.

|
Download:
|
| Fig. 4. Fluorescence polarization competition assays characterizing the binding affinity of dimeric peptides to MDM2; data are expressed as mean ±s.d. (n = 3). | |
{kind=link}
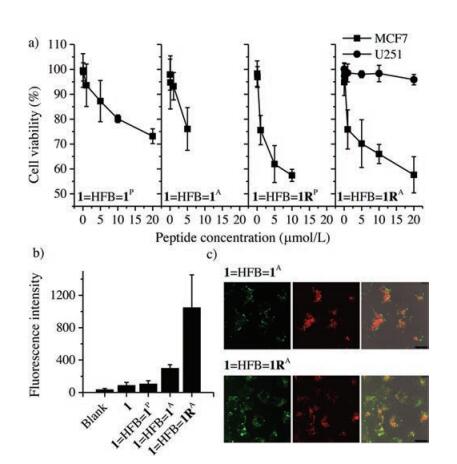
In the light of the high proteolytic stability and affinity of these interchain doubly-bridged α-helical peptides, we then evaluated their biological activity in cells using Cell Counting Kit- 8 [41]. Peptide 1 can block p53-MDM2 binding interaction in cells and activate the p53 pathway to promote apoptosis of cancer cells. MCF7 breast cancer cells expressing wild-type p53 and MDM2 were selected for the examination. Upon incubation of our peptide dimers at concentrations from 0.01 mmol/L to 20 mmol/L with cells, we observed concentration-dependent apoptosis (Fig. 5a), and 1=HFB=1RA displays a stronger cell-killing capability at equivalent concentrations compared to 1=HFB=1P and 1=HFB=1A. To confirm that the peptide-induced apoptosis is closely associated with the p53 activation, we chose a p53 mutant cancer cell line, U251 human glioma cells, for comparison [52]. The cell-killing capability of peptides to U251 cells is obviously weaker than that to MCF7 cells, suggesting that modulation of the intracellular MDM2-p53 interactions should take responsibility for the apoptosis. We further synthesized fluorescent dye (Oregon Green 488 dye)-labeled peptide probes to explore the cellular uptake of dimeric peptides. The cellular uptake of these peptide probes was evaluated by flow cytometry (Fig. 5b). We found that the cellular uptake efficiency of 1=HFB=1RA is significantly higher than other peptides, strongly suggesting the positive effect of the incorporation of positively charged arginines on the enhanced cellular uptake. Confocal fluorescence imaging also confirms that the arginines facilitate the cellular uptake, because cells incubated with 1=HFB=1RA show obviously stronger intracellular fluorescence compare to cells incubated with 1=HFB=1A (Fig. 5c).

|
Download:
|
| Fig. 5. (a) Viability of MCF7 and U251 cells was determined using CCK-8 after incubation with different peptides of various concentrations; data are expressed as mean ± s.d. (n = 3). (b) Flow cytometric analysis of MCF7 cells incubated with 1.0 μmol/L peptide probes. Mean fluorescence intensity per cell was shown as the indicator of cellular uptake efficiency; data are expressed as mean ± s.d. (n = 3). (c) Representative confocal fluorescence images of MCF7 cells exposed to 1.0 μmol/L peptide dimers (top: 1=HFB=1A; bottom: 1=HFB=1RA); LysoTracker Red DND-99 was used to label lysosomes; from left to right: fluorescein channels for peptides, DND channels for lysosomes, an overlay of bright field and fluorescence images. | |
{kind=link}
In summary, we report the design and synthesis of a kind of novel interchain doubly-bridged α-helical peptides by taking advantage of the mutual stabilization of two peptide α-helices via two i and i+7 covalent crosslinkers. These peptides provide a promising platform for developing potent protein binders with high proteolytic stability and affinity. For example, in this work we designed potent inhibitors to the intracellular p53-MDM2 interaction. As many protein-protein interactions take an interfacial α-helix as the binding motif, more protein binders can thus be developed in the future by exploiting our dimeric peptides as a scaffolds.
AcknowledgmentsWe would like to acknowledge the financial support from the National Natural Science Foundation of China (Nos. 21675132 and 21822404), the Program for Changjiang Scholars and Innovative Research Team in University (No. 13036) and the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No. 21521004).
| [1] |
C.J. White, A.K. Yudin, Nat. Chem. 3 (2011) 509-524. DOI:10.1038/nchem.1062 |
| [2] |
W.D. Liu, C.L. Wu, Chin. Chem. Lett. 29 (2018) 1063-1066. DOI:10.1016/j.cclet.2018.03.015 |
| [3] |
G.F. King, Expert Opin. Biol. Ther. 11 (2011) 1469-1484. DOI:10.1517/14712598.2011.621940 |
| [4] |
A.A. Kaspar, J.M. Reichert, Drug Discov. Today 18 (2013) 807-817. DOI:10.1016/j.drudis.2013.05.011 |
| [5] |
R.J. Clark, J. Jensen, S.T. Nevin, et al., Angew. Chem. Int. Ed. 49 (2010) 6545-6548. DOI:10.1002/anie.201000620 |
| [6] |
B.A. Smith, D.S. Daniels, A.E. Coplin, et al., J. Am. Chem. Soc. 130 (2008) 2948-2949. DOI:10.1021/ja800074v |
| [7] |
L. Peraro, J.A. Kritzer, Angew. Chem. Int. Ed. 57 (2018) 11868-11881. DOI:10.1002/anie.201801361 |
| [8] |
T. Okamoto, K. Zobel, A. Fedorova, et al., ACS Chem. Biol. 8 (2013) 297-302. DOI:10.1021/cb3005403 |
| [9] |
Y. Jiang, H.Y. Long, Y.J. Zhu, Y. Zeng, Chin. Chem. Lett. 29 (2018) 1067-1073. DOI:10.1016/j.cclet.2018.05.028 |
| [10] |
G.M. Fang, X.X. Chen, Q.Q. Yang, et al., Chin. Chem. Lett. 29 (2018) 1033-1042. DOI:10.1016/j.cclet.2018.02.002 |
| [11] |
T.A. Hill, N.E. Shepherd, F. Diness, D.P. Fairlie, Angew. Chem. Int. Ed. 53 (2014) 13020-13041. DOI:10.1002/anie.201401058 |
| [12] |
M. Liu, C. Li, M. Pazgier, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 14321-14326. DOI:10.1073/pnas.1008930107 |
| [13] |
Y.X. Wang, D.H.C. Chou, Angew. Chem. Int. Ed. 54 (2015) 10931-10934. DOI:10.1002/anie.201503975 |
| [14] |
Y. Guo, D.M. Sun, F.L. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 14276-14281. DOI:10.1002/anie.201500699 |
| [15] |
G.J.J. Richelle, S. Ori, H. Hiemstra, van Maarseveen J.H., P. Timmerman, Angew. Chem. Int. Ed. 57 (2018) 501-505. DOI:10.1002/anie.201709127 |
| [16] |
W.D. Liu, Y.W. Zheng, X.D. Kong, et al., Angew. Chem. Int. Ed. 56 (2017) 4458-4463. DOI:10.1002/anie.201610942 |
| [17] |
J.H. Wang, M.R. Zha, Q.R. Fei, et al., Chem.-Eur. J. 23 (2017) 15150-15155. DOI:10.1002/chem.201703139 |
| [18] |
J.S. Zheng, S. Tang, Y. Guo, H.N. Chang, L. Liu, ChemBioChem 13 (2012) 542-546. DOI:10.1002/cbic.v13.4 |
| [19] |
C. Xu, J.C. Xu, H. Liu, X.C. Li, Chin. Chem. Lett. 29 (2018) 1119-1122. DOI:10.1016/j.cclet.2018.03.012 |
| [20] |
Y.S. Chang, B. Graves, V. Guerlavais, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) E3445-E3454. DOI:10.1073/pnas.1303002110 |
| [21] |
L. Peraro, Z.J. Zou, K.M. Makwana, et al., J. Am. Chem. Soc. 139 (2017) 7792-7802. DOI:10.1021/jacs.7b01698 |
| [22] |
Y. Wu, Y.H. Li, X. Li, et al., Chem. Sci. 8 (2017) 7368-7373. DOI:10.1039/C7SC02420G |
| [23] |
Y. Tian, J.X. Li, H. Zhao, et al., Chem. Sci. 7 (2016) 3325-3330. DOI:10.1039/C6SC00106H |
| [24] |
Z.P.A. Wang, X.Z.Z. Ding, C.L. Tian, J.S. Zheng, RSC Adv. 6 (2016) 61599-61609. DOI:10.1039/C6RA13976K |
| [25] |
J.S. Appelbaum, J.R. LaRochelle, B.A. Smith, et al., Chem. Biol. 19 (2012) 819-830. DOI:10.1016/j.chembiol.2012.05.022 |
| [26] |
K. Sakagami, T. Masuda, K. Kawano, S. Futaki, Mol. Pharm. 15 (2018) 1332-1340. DOI:10.1021/acs.molpharmaceut.7b01130 |
| [27] |
G.H. Bird, E. Mazzola, Opoku-Nsiah K., et al., Nat. Chem. Biol. 12 (2016) 845-852. DOI:10.1038/nchembio.2153 |
| [28] |
V. Azzarito, K. Long, N.S. Murphy, A.J. Wilson, Nat. Chem. 5 (2013) 161-173. DOI:10.1038/nchem.1568 |
| [29] |
M.L. Stewart, E. Fire, A.E. Keating, L.D. Walensky, Nat. Chem. Biol. 6 (2010) 595-601. DOI:10.1038/nchembio.391 |
| [30] |
H. Jo, N. Meinhardt, Y.B. Wu, et al., J. Am. Chem. Soc. 134 (2012) 17704-17713. DOI:10.1021/ja307599z |
| [31] |
H.N. Hoang, R.W. Driver, R.L. Beyer, et al., Angew. Chem. Int. Ed. 55 (2016) 8275-8279. DOI:10.1002/anie.201602079 |
| [32] |
K. Hu, H. Geng, Q.Z. Zhang, et al., Angew. Chem. Int. Ed. 55 (2016) 8013-8017. DOI:10.1002/anie.201602806 |
| [33] |
A.M. Spokoyny, Y.K. Zou, J.J. Ling, et al., J. Am. Chem. Soc. 135 (2013) 5946-5949. DOI:10.1021/ja400119t |
| [34] |
X. Li, Y. Zou, H.G. Hu, Chin. Chem. Lett. 29 (2018) 1088-1092. DOI:10.1016/j.cclet.2018.01.018 |
| [35] |
L.D. Walensky, A.L. Kung, I. Escher, et al., Science 305 (2004) 1466-1470. DOI:10.1126/science.1099191 |
| [36] |
A. Muppidi, Z.Y. Wang, X.L. Li, J.D. Chen, Q. Lin, Chem. Commun. 47 (2011) 9396-9398. DOI:10.1039/c1cc13320a |
| [37] |
F. Bernal, M. Wade, M. Godes, et al., Cancer Cell 18 (2010) 411-422. DOI:10.1016/j.ccr.2010.10.024 |
| [38] |
M. Wade, Y.C. Li, G.M. Wahl, Nat. Rev. Cancer 13 (2013) 83-96. DOI:10.1038/nrc3430 |
| [39] |
W. Borcherds, F.X. Theillet, A. Katzer, et al., Nat. Chem. Biol. 10 (2014) 1000-1002. DOI:10.1038/nchembio.1668 |
| [40] |
C.J. Brown, S. Lain, C.S. Verma, A.R. Fersht, D.P. Lane, Nat. Rev. Cancer 9 (2009) 862-873. DOI:10.1038/nrc2763 |
| [41] |
Y.Q. Chen, C.Q. Yang, T. Li, et al., Biomacromolecules 16 (2015) 2347-2355. DOI:10.1021/acs.biomac.5b00567 |
| [42] |
S. Aubry, F. Burlina, E. Dupont, et al., FASEB J. 23 (2009) 2956-2967. DOI:10.1096/fj.08-127563 |
| [43] |
W. Gao, T. Li, J.H. Wang, Y.B. Zhao, C.L. Wu, Anal. Chem. 89 (2017) 937-944. DOI:10.1021/acs.analchem.6b04096 |
| [44] |
T. Li, W. Gao, J.J. Liang, et al., Anal. Chem. 89 (2017) 8501-8508. DOI:10.1021/acs.analchem.7b02084 |
| [45] |
S. Jang, S. Hyun, S. Kim, et al., Angew. Chem. Int. Ed. 53 (2014) 10086-10089. DOI:10.1002/anie.201404684 |
| [46] |
C.L. Wu, S. Wang, L. Brulisauer, J.C. Leroux, M.A. Gauthier, Biomacromolecules 14 (2013) 2383-2388. DOI:10.1021/bm400501c |
| [47] |
C.M.B.K. Kourra, N. Cramer, Chem. Sci. 7 (2016) 7007-7012. DOI:10.1039/C6SC02285E |
| [48] |
J. Yang, H. Chen, I.R. Vlahov, J.X. Cheng, P.S. Low, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 13872-13877. DOI:10.1073/pnas.0601455103 |
| [49] |
L. Brulisauer, N. Kathriner, M. Prenrecaj, M.A. Gauthier, J.C. Leroux, Angew. Chem. Int. Ed. 51 (2012) 12454-12458. DOI:10.1002/anie.201207070 |
| [50] |
S. Hyun, J. Na, S.J. Lee, S. Park, J. Yu, ChemBioChem 11 (2010) 767-770. DOI:10.1002/cbic.201000072 |
| [51] |
M. Pazgiera, M. Liu, G.Z. Zou, et al., Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 4665-4670. DOI:10.1073/pnas.0900947106 |
| [52] |
Y.Q. Chen, T. Li, J.G. Li, et al., Org. Biomol. Chem. 15 (2017) 1921-1929. DOI:10.1039/C6OB02786E |