 2019, Vol. 30
2019, Vol. 30 


b CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety and CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China;
c University of Chinese Academy of Sciences, Beijing 100049, China
Discrete nanoparticles (NPs)assembly that combines multiple NP building blocks into a single multifunctional superstructure is a central theme in nanotechnology for a variety of applications [1,2]. Previously, discrete NP superassemblies were constructed by layerby-layer assembly [3], self-limited growth [4]or specific controlover biomolecular motifs [5]. Recently, DNA has attracted great attention as an agent to control the self-assembly of NPs because of the high programmability and biocompatibility [5-15]. DNA-functionalized materials has expanded significantly to broad applications in biosensing, NPs self-assembly, and drug and gene delivery [16-18]. Although some efforts was made to controllably assemble fluorescent quantum dots (QDs) with DNA [11-13], the method was mainly applied to homo-assembly of gold nanoparticles (AuNPs) because the functionalization of DNA is well established for gold surfaces but not generally applicable for many other inorganic NPs [5-10]. Furthermore, DNA mediated self-assembly of NPs tend to require the controlled DNA-functionalization on the surface of NPs, which is time- and cost-consuming and lacks highyields, preventing practical applications of NP superstructures.
Most recently, lanthanide-doped upconversion nanoparticles (UCNPs) become as an exciting new class of nanophosphors because of their unique optical properties (such as conversion of near infrared (NIR) excitation light into shorter wavelength luminescence, tunable multicolor emission, and exceptional photostability) and diverse applications such as bioimaging, biosensing, and therapeutics [19-30]. Although UCNPs-based multifunctional materials have been achieved through direct attachment of other types of NPs [31,32], assembly of UCNPs-based heterostructures is hampered by controlling the binding sites on its surface because they are normally capped with hydrophobic ligands. Recently, we report a novel strategy for regiospecific assembly of DNA-modified gold nanoparticles (DNA-AuNPs) onto UCNPs [14]. Liu et al. further demonstrated assembly of DNA-AuNPs on the surface of diverse nanomaterials to achieve polyvalent DNA heterostructures [15]. Despite of progress made, reducing complexity in the synthesis process and increasing functionality of the obtained heterostructures remain a central theme in the field of DNA-mediated NP assembly.
Here, we present a simple and universal approach for assembly of hetero-nanostructures consisting of UCNPs dendritically decorated with CdTe quantum dots (QDs) (Scheme 1). QDs, also known as nanoscale semiconductor crystals, is another exciting candidate among the various types of potential NP building blocks because of their unique optical and electronic properties, such as bright fluorescence, size-tunable narrow emission, and high photostability [33,34]. QD/UCNPs heterostructures was previously synthesized through a seeded-growth method [35]. The resulted hetero-nanostructures is hydrophobic and limited their biomedical applications. In our design, DNA not only serves as a template to synthesize QDs but also as an agent for NP assembly. Our approach relies on the binding of DNA (on the surface of QDs through coordination interaction) with UCNPs through the strong coordinating interactions between DNA and lanthanide metal ions on the surface. The resulting assembly retains both the biorecognition function of DNA and the optical properties of UCNPs and QDs, allowing targeted dual-modality imaging of cancer cells.

|
Download:
|
| Scheme 1. Schematic showing the assembly of DNA-QD/UCNPs heteronanostructures. | |
{kind=link}
Generation of heterostructures from the NPs was achieved according to Scheme 1 as follows: (1) A hybrid DNA template containing a phosphorothioate domain (purple) was designed for growth of the QD, a linking spacer containing phosphodiester linkages (blue) and one phosphate domain at the end (orange) for the NP assembly. The synthesis of DNA-functionalized CdTe QDs was conducted by using CdCl2 and NaHTe as precursors and mercaptopropionic acid (MPA) as a co-ligand to passivate sites left open by the DNA. Since Cd2+ ions exhibit much higher affinity for sulfur over oxygen binding in nucleotides, the phosphorothioate portion of the sequence would bind to the surface of QD, while the phosphate domain of the DNA sequence remains free [36]. (2) We then synthesized NaYF4:19%Yb/1%Tm UCNPs using oleic acid as the capping agent through a thermal decomposition method, wherein Yb/Tm was codoped to realize visible and ultraviolet upconversion luminescence (UCL) from Tm3+ under near-infrared excitation. The as-synthesized oleic acid-capped UCNPs were transferred into aqueous phase through an acid induced ligand removal process. (3) In a typical NP assembly process, a proper volume of DNA-QDs solution was mixed with a buffer solution of ligand-free UCNPs. This mixture was kept overnight at room temperature with shaking. The resulted DNA-QD/UCNPs assemblies were collected by centrifugation and washed with water to remove unassembled DNA-QDs.
The as-synthesized DNA-QDs has an average diameter of about 3 nm with strong green fluorescence and broad absorption range (Fig. 1a and Fig. S1 in Supporting information). The as-synthesized UCNPs display uniform morphology with mean sizes of approximately 40 nm (Fig. S2 in Supporting information). As shown in Fig. 1b, ligand-free UCNPs are well-dispersed in water without change in shape, suggesting that these nanocrystals have become hydrophilic. Fig. 1c presents representative TEM images of the assynthesized DNA-QD/UCNPs without any further purification, which showed that the resulting material consisted of a UCNP core decorated with CdTe QDs. Detailed information about the structure of the heterogeneous assemblies was further assayed with highangle annular dark-field scanning transmission electron microscopy-energy dispersive X-ray spectroscopy (HAADF-STEM-EDS) elemental mapping (Fig. 1d). Elemental mapping study of F, Y, Cd and Te on a single DNA-QD/UCNPs confirms its core-shell structure in term of both geometrical and compositional distributions. The core-shell distributed elements within the single heterostructure is also confirmed by the EDS line scan profiles (Fig. S3 in Supporting information). The hydrodynamic size of the ligand-free UCNPs changed from 106.3 nm to 128.7 nm after the assembly with DNA-QDs (Fig. 1e), this 22 nm increase in diameter is consistent with the attachment of DNA-QDs onto the UCNPs. Zeta potential measurement showed that ligand-free UCNPs were positively charged (+28.23 mV) and became negatively charged (-13.93 mV) after the assembly (Fig. 1f), further confirming the formation of the heteroassembled nanostructures composed of a UCNP core decorated with DNA-QDs. The obtained NPs were stable in water and HEPES buffer (pH 7.4 and pH 6.0) (Fig. S4 in Supporting information). The formation of the assemblies was controllable by tuning the ratio of the two building blocks (Fig. S5 in Supporting information). Since DNA molecules could bind on the surface of UCNPs through their strong coordination interaction with lanthanide ions [17], the assembly of DNA-QDs on UCNPs in the system could be attribute to this interfacial interaction.

|
Download:
|
| Fig. 1. TEM images of (a) DNA-CdTe QDs, (b) UCNPs and (c) DNA-QD/UCNPs. (d) HAADF-STEM image and corresponding elemental mapping images of DNA-QD/ UCNPs (blue for F, green for Y, red for Cd, and purple for Te). (e) hydrodynamic size and (f) zeta potential of DNA-QD/UCNPs. Scale bar = 50 nm. | |
{kind=link}
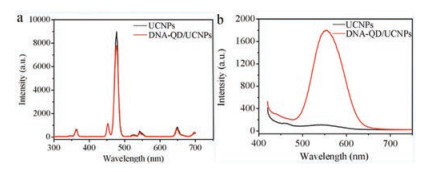
Combining fluorescent nanomaterials with UCNPs has been of great interest due to their various applications, such as multimodal imaging [21]. We investigated the optical properties of the DNA-QD/UCNPs heterostructures. As shown in Fig. 2a, when the ligandfree UCNPs were illuminated with 980 nm continuous wave laser, the Yb3+ ions transfer energy to Tm3+ and results in characteristic emission peaks at ultraviolet (360 nm, 1D2 to 3H6 transition) and visible blue (450 and 475 nm, 1D2 to 3F4 and 1G4 to 3H6, respectively.) (Fig. S6 in Supporting information). As displayed in Fig. 2a and Fig. S7 in Supporting information, the formation of the heterostructures had little influence on the intensity and lifetime of UCL of UCNPs. In addition, the broad band-gap absorption peak (Fig. S8 in Supporting information) and corresponding emission band (548.6 nm) (Fig. 2b) from DNA-QDs were clearly observed for the DNA-QD/UCNPs. Therefore, the assembly enables multifunctional materials because of their combined fluorescence properties of QDs and UCL properties of UCNPs.

|
Download:
|
| Fig. 2. (a) UCL spectra (λex = 980 nm) and (b) fluorescence spectra (λex = 400 nm) of UCNPs and DNA-QD/UCNPs, respectively.of the DNA-QD/UCNPs heterostructures. | |
{kind=link}
It is noteworthy that the strategy resulted in addressable polyvalent DNA modification on the surface of hetero-nanostructures, which may exhibit biorecognition property and thus facilitate targeted bioimaging. We first explored their ability to enter live cells via incubation of the hetero-nanostructures with HeLa cells for 12 h. After washing, the cells subjected to confocal laser scanning microscopy (CLSM) imaging displayed bright NIR-light excited UCL luminescence as well as co- localized fluorescence derived from DNA-QDs (Fig. S9 in Supporting information), demonstrating that the nanoassemblies are able to be internalized without transfection agents. Furthermore, negligible difference in cell viability was observed upon treatment with DNA-QD/UCNPs of different concentrations (Fig.S10 in Supporting information), suggesting the good biocompatibility of the hetero-nanostructures.
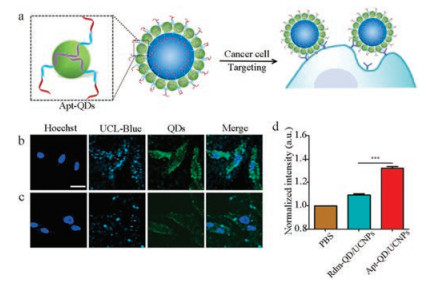
Finally, we explored the use of the strategy to endow heteronanostructures with specific biorecognition capabilities for targeted dual-modality imagingof live cancer cells byintroducing DNA aptamers (Fig. 3a). Aptamers are short single-stranded oligonucleotides that are able to fold into well-defined conformations to bind to various targets with high affinity and specificity [37]. As a proof-of-concept experiment, AS1411, a DNA aptamer that is capable of targeting nucleolin on MDA-MB231 cancer cells [38], was used to as phosphate domain in the DNA template for the synthesis of aptamer-conjugated QDs (Apt-QDs). As shown in Fig. 3b, fluorescence microscopy revealed that MDA-MB231 cells treated with the resulted assembilies displayed both strong upconversion luminescence originated from UCNPs and green fluorescence signals from QDs. The good overlap of the two imaging signals confirms the cellularuptakeof thehetero-nanostructures.In contrast, hetero-nanostructuresmodifiedwith controlDNA showed much weaker internalization by the MDA-MB231 cells (Fig. 3c), confirming the targeting role of the aptamer sequence. Furthermore, flowcytometryanalysis (Fig. 3d) showedthat theintracellular fluorescence intensity of cells incubated with Apt-QD/UCNPs was much higher than that of cells treated with Rdm-QD/UCNPs.

|
Download:
|
| Fig. 3. (a) Schematic showing targeted imaging of cancer cells with Apt-QD/UCNPs. Confocal microscopy images of MDA-MB-231 cells treated with (b) Apt-QD/UCNPs and (c) Rdm-QD/UCNPs. For UCNPs images, λex = 980 nm, λem = 420–500 nm. For DNA-QDs images, λex = 488 nm, λem = 500–600 nm. Scale bar = 20 mm. (d) Flow cytometry quantification of the cellular uptake of Apt-QD/UCNPs and RdmQD/UCNPs. Data are represented as means ± SD, n = 3, ***P < 0.001. | |
{kind=link}
In conclusion, we have devised a simple and general method for the controlled synthesis of multifunctional hetero-nanostructures through DNA-mediated assembly of QDs and UCNPs. The selfregulated mechanism offers high versatility in tailoring the structure of the assemblies. The ready access to nanostructures with multivalent DNA on their surface provides an unparalleled opportunity for multimodal bioimaging. Undoubtedly, the platform combining DNA nanostructures with multifunctional properties has a great potential for biosensing, bioimaging, and drug/ gene delivery.
AcknowledgmentsThis work was supported financially by the National Natural Science Foundation of China (Nos. 21822401, 21771044), and the Young Thousand Talented Program.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.03.022.
| [1] |
L. Wang, L. Xu, H. Kuang, et al., Acc. Chem. Res. 45 (2012) 1916-1926. DOI:10.1021/ar200305f |
| [2] |
Z. Nie, A. Petukhova, E. Kumacheva, Nat. Nanotechnol. 5 (2010) 15-25. DOI:10.1038/nnano.2009.453 |
| [3] |
S. Srivastava, N.A. Kotov, Acc. Chem. Res. 41 (2008) 1831-1841. DOI:10.1021/ar8001377 |
| [4] |
Y. He, T. Ye, A.E. Ribbe, et al., J. Am. Chem. Soc. 133 (2011) 1742-1744. DOI:10.1021/ja1060092 |
| [5] |
L. Xu, H. Kuang, C. Xu, et al., J. Am. Chem. Soc. 134 (2012) 1699-1709. DOI:10.1021/ja2088713 |
| [6] |
Y. Xia, T. Nguyen, M. Yang, et al., Nat. Nanotechnol. 7 (2012) 479. DOI:10.1038/nnano.2012.106 |
| [7] |
L.H. Tan, H. Xing, Y. Lu, Acc. Chem. Res. 47 (2014) 1881-1890. DOI:10.1021/ar500081k |
| [8] |
C.A. Mirkin, R.L. Letsinger, R.C. Mucic, et al., Nature 382 (1996) 607-609. DOI:10.1038/382607a0 |
| [9] |
M.R. Jones, N.C. Seeman, C.A. Mirkin, Science 347 (2015) 1260901. DOI:10.1126/science.1260901 |
| [10] |
L.Y.T. Chou, K. Zagorovsky, W.C.W. Chan, Nat. Nanotechnol. 9 (2014) 148-155. DOI:10.1038/nnano.2013.309 |
| [11] |
R. Schreiber, J. Do, E.M. Roller, et al., Nat. Nanotechnol. 9 (2014) 74-78. DOI:10.1038/nnano.2013.253 |
| [12] |
Y. Zhang, F. Lu, K.G. Yager, et al., Nat. Nanotechnol. 8 (2013) 865-872. DOI:10.1038/nnano.2013.209 |
| [13] |
R. Wang, C. Nuckolls, S.J. Wind, Angew. Chem. Int. Ed. 51 (2012) 11325-11327. DOI:10.1002/anie.201206389 |
| [14] |
L.L. Li, Y. Lu, J. Am. Chem. Soc. 137 (2015) 5272-5275. DOI:10.1021/jacs.5b01092 |
| [15] |
B. Liu, Z. Huang, J. Liu, Angew. Chem. Int. Ed. 57 (2018) 9439-9442. DOI:10.1002/anie.201805532 |
| [16] |
W. Zhou, R. Saran, J. Liu, Chem. Rev. 117 (2017) 8272-8325. DOI:10.1021/acs.chemrev.7b00063 |
| [17] |
L.L. Li, P. Wu, K. Hwang, et al., J. Am. Chem. Soc. 135 (2013) 2411-2414. DOI:10.1021/ja310432u |
| [18] |
M. Li, C. Wang, Z. Di, et al., Angew. Chem. Int. Ed. 58 (2019) 1350-1354. DOI:10.1002/anie.201810735 |
| [19] |
F. Wang, Y. Han, C.S. Lim, et al., Nature 463 (2010) 1061-1065. DOI:10.1038/nature08777 |
| [20] |
M. Haase, H. Schäfer, Angew. Chem. Int. Ed. 50 (2011) 5808-5829. DOI:10.1002/anie.v50.26 |
| [21] |
H. Dong, S.R. Du, X.Y. Zheng, et al., Chem. Rev. 115 (2015) 10725-10815. DOI:10.1021/acs.chemrev.5b00091 |
| [22] |
N.M. Idris, M.K. Gnanasammandhan, J. Zhang, et al., Nat. Med. 18 (2012) 1580-1585. DOI:10.1038/nm.2933 |
| [23] |
W. Ma, P. Fu, M. Sun, et al., J. Am. Chem. Soc. 139 (2017) 11752-11759. DOI:10.1021/jacs.7b03617 |
| [24] |
J. Zhou, Q. Liu, W. Feng, et al., Chem. Rev. 115 (2015) 395-465. DOI:10.1021/cr400478f |
| [25] |
J. Zhao, J. Gao, W. Xue, et al., J. Am. Chem. Soc. 140 (2018) 578-581. DOI:10.1021/jacs.7b11161 |
| [26] |
B. Zhou, B. Shi, D. Jin, et al., Nat. Nanotechnol. 10 (2015) 924-936. DOI:10.1038/nnano.2015.251 |
| [27] |
R. Shi, X. Ling, X. Li, et al., Nanoscale 9 (2017) 13739-13746. DOI:10.1039/C7NR04877G |
| [28] |
Y. Wang, G. Yang, Y. Wang, et al., Nanoscale 9 (2017) 4759-4769. DOI:10.1039/C6NR09030C |
| [29] |
Z. Li, H. Liu, H. Li, et al., Sens. Actuators B Chem. 280 (2019) 94-101. DOI:10.1016/j.snb.2018.10.057 |
| [30] |
G. Tian, X. Zhang, Z. Gu, et al., Adv. Mater. 27 (2015) 7692-7712. DOI:10.1002/adma.201503280 |
| [31] |
Y. Zhang, L. Zhang, R. Deng, et al., J. Am. Chem. Soc. 136 (2014) 4893-4896. DOI:10.1021/ja5013646 |
| [32] |
Y. Li, Z. Di, J. Gao, et al., J. Am. Chem. Soc. 139 (2017) 13804-13810. DOI:10.1021/jacs.7b07302 |
| [33] |
K. Ding, L. Jing, C. Liu, et al., Biomaterials 35 (2014) 1608-1617. DOI:10.1016/j.biomaterials.2013.10.078 |
| [34] |
G. Tikhomirov, S. Hoogland, P.E. Lee, et al., Nat. Nanotechnol. 6 (2011) 485-490. DOI:10.1038/nnano.2011.100 |
| [35] |
C. Yan, A. Dadvand, F. Rosei, et al., J. Am. Chem. Soc. 132 (2010) 8868-8869. DOI:10.1021/ja103743t |
| [36] |
N. Ma, E.H. Sargent, S.O. Kelley, Nat. Nanotechnol. 4 (2009) 121-125. DOI:10.1038/nnano.2008.373 |
| [37] |
J.H. Lee, M.V. Yigit, D. Mazumdar, et al., Adv. Drug Deliv. Rev. 62 (2010) 592-605. DOI:10.1016/j.addr.2010.03.003 |
| [38] |
S. Soundararajan, W. Chen, E.K. Spicer, et al., Cancer Res. 68 (2008) 2358-2365. DOI:10.1158/0008-5472.CAN-07-5723 |