 2019, Vol. 30
2019, Vol. 30 


b College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China;
c Laboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China
Transition metals are used extensively to assist C─C bond forming reactions, and this has attracted broad interest from organic chemists for preparing new drug molecules, functional materials, industrial raw materials, and natural products [1-7]. Various named reactions using transition metals as catalysts or reagents are widely applied in synthetic chemistry [8-11]. The Pauson-Khand reaction is perhaps the most unique name organometallic reaction, which can efficiently and simultaneously construct three C─C bonds [12-18].
The intermolecular Pauson-Khand reaction can be used to construct five-membered cyclopentenone type products through a three-component reaction with an alkene, alkyne, and carbon monoxide (CO). Alternatively, the intramolecular Pauson-Khand reaction with 1, 6- or 1, 7-enyne substrates can yield bicyclo [3.3.0] or [4.3.0] enone derivatives [19-24]. Especially, the Pauson-Khand reaction is widely used for efficiently constructing carbon skeletons in the total synthesis of complicated natural products and organic molecules [25-31]. The first report of the Pauson-Khand reaction by Khand, Knox, Pauson, and Watts employed hexacarbonyldicobalt(0) complexes of alkynes as a carbonyl source and to assist the construction of the cyclopentene [32, 33]. Various organometallic catalysts involving Co [34-37], Rh [38-40], Pd [41-43], Ti [44, 45], Zr [46, 47], Ni [48-50], Ru [51-53], and Ir [54, 55] have since been developed, and often exhibit individual reactivities and substrate compatibilities. However, stoichiometric Co2(CO)8 is still the most widely used for the Pauson-Khand reaction.
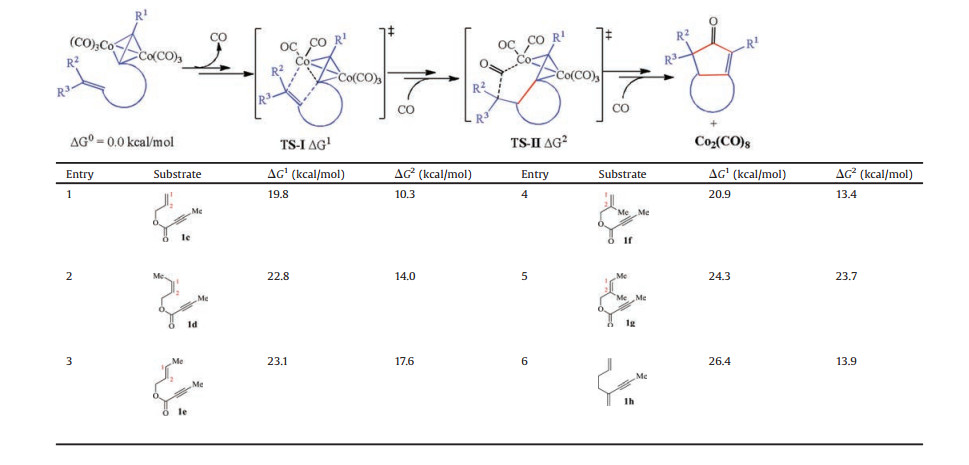
The well-accepted reaction mechanism for the Co2(CO)8-mediated Pauson-Khand reaction was proposed by Magnus in 1985 and supported by density functional theory (DFT) calculations in 2001 [56, 57]. The reaction starts from reversible ligand exchange by the alkyne to form an alkyne-coordinated Co2(CO)6 species. Subsequent oxidative cyclization with the olefin forms a five-membered cobaltacycle intermediate. The sequential carbonyl insertion into vinylcobalt and reductive elimination affords the corresponding cyclopentenone complex. Following this idea, a series of theoretical and experimental studies have been carried out in attempt to evaluate the reactivity and selectivity of the Co2(CO)8-mediated Pauson-Khand reaction [58-62].
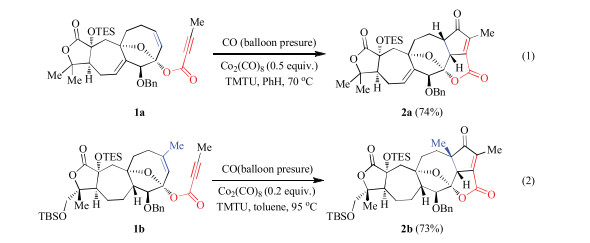
Recently, some of us reported a series of Co2(CO)8-mediated Pauson-Khand reactions for constructing bicyclo-skeletons in the total synthesis of natural products (Scheme 1) [63, 64]. In these reactions, the cyclooctene derivative can be used as a substrate for the Pauson-Khand reaction under relatively mild reaction conditions. The reaction also proceeded smoothly when the methyl substituted olefin component 1b was employed. However, the reason why the reaction could proceed at mild conditions and what controls the reactivity remain unclear. Herein, DFT calculations are employed to explore the influence of the substrate on the reactivity of the Pauson-Khand reaction.

|
Download:
|
| Scheme 1. Co2(CO)8-mediated intramolecular Pauson-Khand reaction of cyclooctene derivatives. | |
All the DFT calculations were carried out with the GAUSSIAN 09 series of programs [65]. DFT method B3-LYP [66, 67] with a standard 6–31G(d) basis set (SDD [68, 69] basis set for Co) was used for geometry optimizations. Harmonic frequency calculations were performed for all stationary points to confirm them as a local minima or transition structures and to derive the thermochemical corrections for the enthalpies and free energies. M11-L functional [70-72] with basis set 6-311+G(d, p) (SDD basis set for Co) was employed to calculate the solvation single point energies to give more accurate energy information. The solvent effects were considered by single point calculations on the gas-phase stationary points with a SMD continuum solvation model [73]. The energies given in this work are M11-L calculated Gibbs free energies in solvent.
We initially calculated the free energy profiles for the Co2(CO)8-mediated intramolecular Pauson-Khand reaction of enyne 1a, and the results are summarized in Fig. 1. Enyne 1a reacts with Co2(CO)8 to form the Co2/acetylene complex CP1 with the release of two molecules of CO, which is exergonic by 4.6 kcal/mol. The intramolecular coordination of the alkene moiety to the coordinatively unsaturated complex CP1 then proceeds to give the thermodynamically unfavorable complex CP2 with the release of one molecule of CO. Subsequent alkene insertion into the Co─C bond via transition state TS-1 leads to the reversible generation of cobaltacycle intermediate CP3. The free energy span of this step is determined to be 6.7 kcal/mol. Staring from intermediate CP3, the coordination of CO to Co center generates the thermodynamically favorable intermediate CP4, with a free energy decrease of 4.7 kcal/mol. Subsequent CO insertion occurs via transition state TS2 with a free energy barrier of 14.3 kcal/mol, forming intermediate CP5. Further CO coordination of CP5 is followed by rapid reductive elimination to deliver complex CP6 through transition state TS3. The free energy barrier for this step is only 8.4 kcal/mol. The cyclic product 2a is then released from intermediate CP6 with the concomitant formation of the Co2(CO)8 complex with a significant exotherm.

|
Download:
|
| Fig. 1. Free energy profiles for the Co2(CO)8-mediated intramolecular Pauson-Khand reaction of 1a. Values are given in kcal/mol and represent the relative free energies calculated by the M11-L method in benzene. Bond distances are in angstroms (Å). | |
The overall activation free energy of the reaction pathway is determined to be 16.7 kcal/mol, which suggests that this reaction proceeds smoothly under relatively mild conditions. The calculation results suggest that CO insertion is the rate-determining step for the overall reaction pathway, while alkene insertion leading to the cobaltacycle was identified as the rate-determining step in previous reports of other Pauson-Khand reaction systems. The reduced activation free energy for alkene insertion in this reaction may be attributed to two reasons. The ester group in close proximity to the C─C triple bond serves as an activating group to enhance the reactivity of the alkyne moiety [74]. Also, steric hindrance during alkene insertion is reduced when using the cyclooctene derivative, compared with chain olefin reactants.
In a previous experimental study, the Pauson-Khand reaction proceeded smoothly when the methyl substituted olefin component 1b was employed. Thus, we employed DFT calculations to investigate the Pauson-Khand reaction of 1b, to identify the reactivity effected by the substituted methyl group. As shown in Fig. 2, ligand exchange of the Co2(CO)8 complex and 1a gives intermediate CP7 and the concomitant release of two molecules of CO, with a free energy decrease of 3.9 kcal/mol. The intramolecular coordination of the alkene moiety to the coordinatively unsaturated complex CP7 then proceeds to give the thermodynamically unfavorable complex CP8 with the release of one molecule of CO. Subsequent alkene insertion into the Co-C bond via transition state TS-4 leads to the reversible generation of cobaltacycle intermediate CP9. The activation free energy of this step is determined to be 17.5 kcal/mol (CP7→TS4), which is 2.3 kcal/mol higher than that of the step involving reactant 1a (CP1→TS1). The enhanced activation free energy in the alkene insertion step may be due to steric hindrance introduced by the substituted methyl group.

|
Download:
|
| Fig. 2. Free energy profiles for the Co2(CO)8-mediated intramolecular Pauson-Khand reaction of 1b. Values are given in kcal/mol and represent the relative free energies calculated by the M11-L method in toluene. Bond distances are in angstroms (Å). | |
The coordination of CO to the Co center of intermediate CP9 generates intermediate CP10. CO insertion into the Co-C bond then occurs via transition state TS5 with a free energy barrier of 9.9 kcal/mol, resulting in the acyl-Co intermediate CP11. Under the assistance of CO coordination, subsequent C─C bond reductive elimination occurs rapidly to deliver the product coordinated intermediate CP12. The cyclic product 2b is then released from intermediate CP12 with the concomitant formation of the Co2(CO)8 complex. Unexpectedly, alkene insertion leading to the cobaltacycle is identified as the rate-determining step when the methyl substituted olefin component 1b is employed. The different rate-determining steps indicated by the calculations may be caused by the steric hindrance induced by the substituted methyl group, which increases the activation free energy of the alkene insertion step [75].
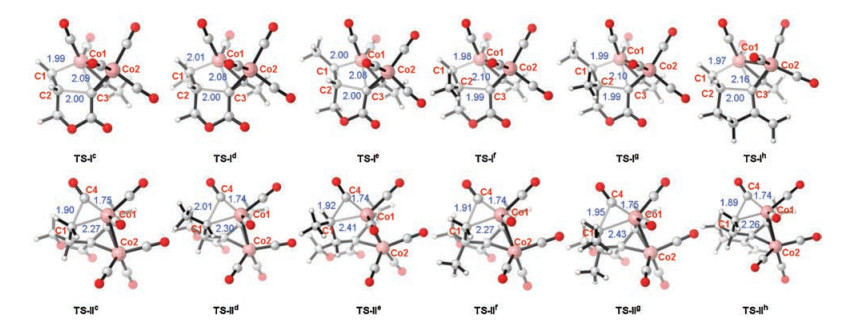
To further assess the reactivity of different substrates in the intramolecular Pauson-Khand reaction, we calculated free energy profiles for the reaction pathways for reactants 1c–h (details see Supporting information). Aforementioned results indicate that the alkene insertion or CO insertion step could be considered as the rate-determining step for the overall reaction pathway, while the subsequent reductive elimination is rapid. The computational results for the two key steps are summarized in Table 1. The resting state hexacarbonyldicobalt(0) complexes of the alkynes are used as the relative zero points. △G1 and △G2 represent the relative free energy of TS-Ⅰ and TS-Ⅱ for the alkene insertion or CO insertion step, respectively. The terminal alkene 1c is employed to evaluate the reactivity of the chain alkene (Entry 1). The activation free energy of the alkene insertion step via transition state TS-Ⅰc is determined to be 19.8 kcal/mol, which is 4.6 kcal/mol higher than that of the step involving the cyclooctene reactant 1a (CP1→TS1). This indicates that steric hindrance during alkene insertion could be significantly reduced by using the cyclooctene reactant. Meanwhile, the alkene insertion leading to the cobaltacycle is identified as the rate-determining step. The activation free energies of the alkene insertion steps are 22.8 (TS-Ⅰd) and 23.1 kcal/mol when the internal alkenes 1d and 1c are used, respectively (entries 2 and 3, respectively). The enhanced activation free energy may be due to steric hindrance introduced by the substituted methyl group. The activation free energies of the CO insertion steps for 1d and 1e are noticeably increased. The increasing C1-Co1 bond lengths in TS-Ⅱd and TS-Ⅱe (2.30 Å and 2.41 Å, respectively) demonstrate the steric hindrance induced by the substituted methyl group (Fig. 3). In contrast, the cis alkene 1d exhibits higher reactivity. For the C2 position methyl-substituted 1f, the activation free energy of the alkene insertion step is slightly higher than that of 1c (entry 4). This suggests that the substituted methyl group at the C2 position induces less steric hindrance compared with 1d and 1e. The activation free energies of the alkene insertion and CO insertion steps are 24.3 kcal/mol and 23.7 kcal/mol, respectively, when 1, 2-dimethyl substituted 1g is employed. In addition, when the O atoms are replaced by C atoms in 1h, the activation free energy of the alkene insertion step significantly increases. This demonstrates that the ester group in close proximity to the C─C triple bond serves as an activating group to enhance the reactivity. Moreover, the tether effects of alkene insertion step were also considered for this intramolecular Pauson-Khand reaction [76-78]. Calculated results indicate that tether effect of the tether involving ester moiety (1c) is significantly larger than the all carbon skeleton (1h).
|
|
Table 1 Substituent effect for the Co2(CO)8-mediated intramolecular Pauson-Khand reaction. Values are given in kcal/mol and represent the relative free energies calculated by the M11-L method in toluene. |
{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Fig. 3. Optimized structures of the transition states in the alkene insertion and CO insertion steps. Bond distances are given in angstroms (Å). | |
{kind=link}
DFT calculations were employed to explore the influence of the substrate on the reactivity of the Co2(CO)8-mediated intramolecular Pauson-Khand reaction, which is commonly used in the total synthesis of natural products with reasonable reaction rates and yields. For the Pauson-Khand reaction involving cyclooctene derivative 1a, CO insertion is considered to be the rate-determining step for the overall reaction pathway. This differs to previous theoretical and experimental studies for Pauson-Khand reactions with regular enynes. The reduced activation free energy for the alkene insertion in this reaction is attributed to: ⅰ) the ester group in close proximity to the C─C triple bond enhancing the reactivity of the alkyne moiety; ⅱ) the lower steric hindrance during alkene insertion when using the cyclooctene derivative. The effect of the substituent on the Co2(CO)8-mediated intramolecular Pauson-Khand reaction was then investigated. Internal alkenes exhibit lower reactivity than terminal alkenes because of the steric hindrance introduced by the substituted groups. The cis internal alkene exhibits higher reactivity than the trans internal alkene. The ester group in close proximity to the C─C triple bond significantly enhances the reactivity. These findings provide a theoretical guide for designing and carrying out Co2(CO)8-mediated Pauson-Khand reactions.
AcknowledgmentsWe are thankful for the project (Nos. 2018CDYJSY0055, 2018CDXZ0002, 106112017CDJXY220007) supported by the Fundamental Research Funds for the Central Universities (Chongqing University). This project was also supported by the National Natural Science Foundation of China (Nos. 21772020 and 21822303).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.03.024.
| [1] |
M. Lautens, W. Klute, W. Tam, Chem. Rev. 96 (1996) 49-92. DOI:10.1021/cr950016l |
| [2] |
C. Liu, H. Zhang, W. Shi, A. Lei, Chem. Rev. 111 (2011) 1780-1824. DOI:10.1021/cr100379j |
| [3] |
T. Mesganaw, J.A. Ellman, Org. Process Res. Dev. 18 (2014) 1097-1104. DOI:10.1021/op500224x |
| [4] |
M. Rubin, M. Rubina, V. Gevorgyan, Chem. Rev. 107 (2007) 3117-3179. DOI:10.1021/cr050988l |
| [5] |
N. Weding, M. Hapke, Chem. Soc. Rev. 40 (2011) 4525-4538. DOI:10.1039/c0cs00189a |
| [6] |
J. Wencel-Delord, F. Glorius, Nat. Chem. 5 (2013) 369-375. DOI:10.1038/nchem.1607 |
| [7] |
J. Yamaguchi, A.D. Yamaguchi, K. Itami, Angew. Chem. Int. Ed. 51 (2012) 8960-9009. DOI:10.1002/anie.201201666 |
| [8] |
K. Takao, R. Munakata, K. Tadano, Chem. Rev. 105 (2005) 4779-4807. DOI:10.1021/cr040632u |
| [9] |
J. Le Bras, J. Muzart, Chem. Rev. 111 (2011) 1170-1214. DOI:10.1021/cr100209d |
| [10] |
J.A. Olson, K.M. Shea, Acc. Chem. Res. 44 (2011) 311-321. DOI:10.1021/ar100114m |
| [11] |
X. Jiang, C. Feng, G. Lu, X. Huang, Sci. China Chem. 58 (2015) 1695-1709. DOI:10.1007/s11426-015-5447-1 |
| [12] |
H.W. Frühauf, Chem. Rev. 97 (1997) 523-596. DOI:10.1021/cr941164z |
| [13] |
O. Geis, H.G. Schmalz, Angew. Chem. Int. Ed. 37 (1998) 911-914. |
| [14] |
K.M. Brummond, J.L. Kent, Tetrahedron 56 (2000) 3263-3283. DOI:10.1016/S0040-4020(00)00148-4 |
| [15] |
S.E. Gibson, A. Stevenazzi, Angew. Chem. Int. Ed. 42 (2003) 1800-1810. DOI:10.1002/anie.200200547 |
| [16] |
S.E. Gibson, N. Mainolfi, Angew. Chem. Int. Ed. 44 (2005) 3022-3037. |
| [17] |
T. Shibata, Adv. Synth. Catal. 348 (2006) 2328-2336. |
| [18] |
J.D. Ricker, L.M. Geary, Top. Catal. 60 (2017) 609-619. DOI:10.1007/s11244-017-0741-0 |
| [19] |
J. Castro, A. Moyano, M.A. Pericàs, A. Riera, A.E. Greene, Tetrahedron Asymmetry 5 (1994) 307-310. DOI:10.1016/S0957-4166(00)86192-3 |
| [20] |
C. Mukai, F. Inagaki, T. Yoshida, et al., J. Org. Chem. 70 (2005) 7159-7171. DOI:10.1021/jo050770z |
| [21] |
Y. Tang, L. Deng, Y. Zhang, et al., Org. Lett. 7 (2005) 593-595. DOI:10.1021/ol047651a |
| [22] |
C. Mukai, T. Yoshida, M. Sorimachi, A. Odani, Org. Lett. 8 (2006) 83-86. DOI:10.1021/ol052562z |
| [23] |
Q. Liu, G. Yue, N. Wu, et al., Angew. Chem. Int. Ed. 51 (2012) 12072-12076. DOI:10.1002/anie.201206705 |
| [24] |
E. Cristóbal-Lecina, A.R. Costantino, A. Grabulosa, A. Riera, X. Verdaguer, Organometallics 34 (2015) 4989-4993. DOI:10.1021/acs.organomet.5b00576 |
| [25] |
T.W. Sun, W.W. Ren, Q. Xiao, et al., Chem. -Asian J. 7 (2012) 2321-2333. DOI:10.1002/asia.201200363 |
| [26] |
D.R. Williams, A.A. Shah, J. Am. Chem. Soc. 136 (2014) 8829-8836. DOI:10.1021/ja5043462 |
| [27] |
Y. Zhang, J. Gong, Z. Yang, Chem. Rec. 14 (2014) 606-622. DOI:10.1002/tcr.v14.4 |
| [28] |
K.V. Chuang, C. Xu, S.E. Reisman, Science 353 (2016) 912-915. DOI:10.1126/science.aag1028 |
| [29] |
C. Lv, X. Yan, Q. Tu, et al., Angew. Chem. Int. Ed. 55 (2016) 7539-7543. DOI:10.1002/anie.201602783 |
| [30] |
A. Cabre, H. Khaizourane, M. Garcon, X. Verdaguer, A. Riera, Org. Lett. 20 (2018) 3953-3957. DOI:10.1021/acs.orglett.8b01525 |
| [31] |
Z. Huang, J. Huang, Y. Qu, et al., Angew. Chem. Int. Ed. 57 (2018) 8744-8748. DOI:10.1002/anie.v57.28 |
| [32] |
I.U. Khand, G.R. Knox, P.L. Pauson, W.E. Watts, J. Chem. Soc. Perkin Trans 1 (1973) 975. |
| [33] |
I.U. Khand, G.R. Knox, P.L. Pauson, W.E. Watts, M.I Foreman, J. Chem. Soc. Perkin Trans 1 (1973) 977. |
| [34] |
J.L. Muller, A. Rickers, W. Leitner, Adv. Synth. Catal. 349 (2007) 287-291. |
| [35] |
D.R. Hartline, M. Zeller, C. Uyeda, Angew. Chem. Int. Ed. 55 (2016) 6084-6087. DOI:10.1002/anie.201601784 |
| [36] |
J. Garcia-Lacuna, G. Dominguez, J. Blanco-Urgoiti, J. Perez-Castells, Chem. Commun. 53 (2017) 4014-4017. DOI:10.1039/C7CC01749A |
| [37] |
Z. Zhang, Y. Li, D. Zhao, et al., Chem.-Eur. J. 23 (2017) 1258-1262. DOI:10.1002/chem.201605438 |
| [38] |
F.Y. Kwong, H.W. Lee, L. Qiu, et al., Adv. Synth. Catal. 347 (2005) 1750-1754. |
| [39] |
K.M. Brummond, M.M. Davis, C. Huang, J. Org. Chem. 74 (2009) 8314-8320. DOI:10.1021/jo901459t |
| [40] |
L.C. Burrows, L.T. Jesikiewicz, G. Lu, et al., J. Am. Chem. Soc. 139 (2017) 15022-15032. DOI:10.1021/jacs.7b07121 |
| [41] |
Y. Tang, L. Deng, Y. Zhang, et al., Org. Lett. 7 (2005) 1657-1659. DOI:10.1021/ol050410y |
| [42] |
Z. Yang, A. Lei, J.H. Chen, et al., Synthesis 39 (2007) 2565-2570. |
| [43] |
Y. Lan, L. Deng, J. Liu, et al., J. Org. Chem. 74 (2009) 5049-5058. DOI:10.1021/jo900919v |
| [44] |
F.A. Hicks, S.L. Buchwald, J. Am. Chem. Soc. 118 (1996) 11688-11689. DOI:10.1021/ja9630452 |
| [45] |
F.A. Hicks, N.M. Kablaoui, S.L. Buchwald, J. Am. Chem. Soc. 118 (1996) 9450-9451. DOI:10.1021/ja9621509 |
| [46] |
E. Negishi, S.J. Holmes, J.M. Tour, J.A. Miller, J. Am. Chem. Soc. 107 (1985) 2568-2569. DOI:10.1021/ja00294a071 |
| [47] |
G. Agnel, E. Negishi, J. Am. Chem. Soc. 113 (1991) 7424-7426. DOI:10.1021/ja00019a051 |
| [48] |
K. Tamao, K. Kobayashi, Y. Ito, J. Am. Chem. Soc. 110 (1988) 1286-1288. DOI:10.1021/ja00212a045 |
| [49] |
M. Zhang, S.L. Buchwald, J. Org. Chem. 61 (1996) 4498-4499. DOI:10.1021/jo960410z |
| [50] |
Y. Hoshimoto, K. Ashida, Y. Sasaoka, et al., Angew. Chem. Int. Ed. 56 (2017) 8206-8210. DOI:10.1002/anie.v56.28 |
| [51] |
T. Kondo, N. Suzuki, T. Okada, Ta. Mitsudo, J. Am. Chem. Soc. 119 (1997) 6187-6188. DOI:10.1021/ja970793y |
| [52] |
K. Itami, K. Mitsudo, K. Fujita, Y. Ohashi, J. Yoshida, J. Am. Chem. Soc. 126 (2004) 11058-11066. DOI:10.1021/ja047484+ |
| [53] |
C. Wang, Y.D. Wu, Organometallics 27 (2008) 6152-6162. DOI:10.1021/om8004178 |
| [54] |
T. Shibata, K. Takagi, J. Am. Chem. Soc. 122 (2000) 9852-9853. DOI:10.1021/ja000899k |
| [55] |
F.Y. Kwong, H.W. Lee, W.H. Lam, L. Qiu, A.S.C. Chan, Tetrahedron Asymmetry 17 (2006) 1238-1252. DOI:10.1016/j.tetasy.2006.03.036 |
| [56] |
P. Magnus, L.M. Principe, Tetrahedron Lett. 26 (1985) 4851-4854. DOI:10.1016/S0040-4039(00)94968-2 |
| [57] |
M. Yamanaka, E. Nakamura, J. Am. Chem. Soc. 123 (2001) 1703-1708. DOI:10.1021/ja005565+ |
| [58] |
T. Fjermestad, M.A. Pericas, F. Maseras, Chem.-Eur. J. 17 (2011) 10050-10057. DOI:10.1002/chem.v17.36 |
| [59] |
D. Lesage, A. Milet, A. Memboeuf, et al., Angew. Chem. Int. Ed. 53 (2014) 1939-1942. DOI:10.1002/anie.201307745 |
| [60] |
S. Liu, H. Shen, Z. Yu, et al., Organometallics 33 (2014) 6282-6285. DOI:10.1021/om500840q |
| [61] |
A.M. Rodriguez, P. Prieto, Tetrahedron 72 (2016) 7443-7448. DOI:10.1016/j.tet.2016.09.048 |
| [62] |
J.P. Martinez, M. Vizuete, L.M. Arellano, et al., Nanoscale 10 (2018) 15078-15089. DOI:10.1039/C8NR03480J |
| [63] |
Q. Xiao, W.W. Ren, Z.X. Chen, et al., Angew. Chem. Int. Ed. 50 (2011) 7373-7377. DOI:10.1002/anie.v50.32 |
| [64] |
D.D. Liu, T.W. Sun, K.Y. Wang, et al., J. Am. Chem. Soc. 139 (2017) 5732-5735. DOI:10.1021/jacs.7b02561 |
| [65] |
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013.
|
| [66] |
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785. DOI:10.1103/PhysRevB.37.785 |
| [67] |
A.D. Becke, J. Chem. Phys 98 (1993) 5648. DOI:10.1063/1.464913 |
| [68] |
M. Dolg, U. Wedig, H. Stoll, H. Preuss, J. Chem. Phys 86 (1987) 866.. DOI:10.1063/1.452288 |
| [69] |
M. Dolg, H. Stoll, H. Preuss, J. Chem. Phys 90 (1989) 1730. DOI:10.1063/1.456066 |
| [70] |
Y. Zhao, H.T. Ng, R. Peverati, D.G. Truhlar, J. Chem. Theory Comput. 8 (2012) 2824-2834. DOI:10.1021/ct300457c |
| [71] |
R. Peverati, D.G. Truhlar, J. Phys. Chem. Lett. 3 (2012) 117-124. DOI:10.1021/jz201525m |
| [72] |
R. Peverati, D.G. Truhlar, Phys. Chem. Chem. Phys. 14 (2012) 11363-11370. DOI:10.1039/c2cp41295k |
| [73] |
A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 113 (2009) 6378-6396. DOI:10.1021/jp810292n |
| [74] |
E. Fager-Jokela, M. Muuronen, H. Khaizourane, et al., J. Org. Chem. 79 (2014) 10999-11010. DOI:10.1021/jo502035t |
| [75] |
T.J. de Bruin, A. Milet, A.E. Greene, Y. Gimbert, J. Org. Chem. 69 (2004) 1075-1080. DOI:10.1021/jo035294w |
| [76] |
X. Hong, Y. Liang, M. Brewer, K.N. Houk, Org. Lett. 16 (2014) 4260-4263. DOI:10.1021/ol501958s |
| [77] |
E.H. Krenske, E.C. Davison, I.T. Forbes, et al., J. Am. Chem. Soc. 134 (2012) 2434-2441. DOI:10.1021/ja211568k |
| [78] |
E.H. Krenske, E.W. Perry, S.V. Jerome, et al., Org. Lett. 14 (2012) 3016-3019. DOI:10.1021/ol301083q |