 2019, Vol. 30
2019, Vol. 30 


b Research Center for Environmental Science & Technology, Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 611731, China;
c 3The Center of New Energy Materials and Technology, School of Materials Science and Engineering, Southwest Petroleum University, Chengdu 610500, China;
d College of Architecture and Environment, Sichuan University, Chengdu 610065, China
The enhanced photocatalytic activity of phosphate/potassium co-functionalized carbon nitride can be attributed to the promoted reactants activation capacity, the decreased of carriers recombination, and the construction of electronic channels between CN layers.
The air pollution caused by the excessive consumption of fossil fuels is one of the global challenges, especially for developing countries [1-5]. NO, as a typical air pollutant, is one of the main causes for acid rain, photochemical smog and other environmental problems [6]. Hence, the development of NO purification technology is highly desirable for air pollution control. In recent years, the photocatalysis has become one of the most promising technologies in environment remediation. Numerous semiconductor photocatalysts (for example, TiO2, ZnO, Ga2O3 and Bi2O2CO3) have been reported [7-14]. Nevertheless, these photocatalysts can only be excited by UV light because of the wide band gap. Consequently, the photocatalysts working under visible light should be developed. The metal-free visible light driven g-C3N4 has been widely studied as photocatalysts with wide application in splitting water to produce H2, decomposition of organic pollutant, and carbon dioxide reduction under visible light [15-20]. Unfortunately, g-C3N4 suffers from high recombination rate and low reactants activation efficiency, which limit its practical application [21-24].
Great efforts have been devoted to improve the photocatalytic efficiency of g-C3N4 over past few years, including surface modification, electronic structure modulation, heterostructure construction, nanostructure design, and metal or non-metal doping [23, 25-35]. With regard to surface modification, it could significantly affect on the photocatalytic process by altering the charge transfer pathways at the catalyst surface [36, 37]. It is wellknown that surface modification of TiO2 with inorganic nonmetal redox-inert anions, such as [PO4]3-, [SO4]2- and F-, can greatly improve the photocatalytic activity [38-44]. Liu et al. prepared modified g-C3N4 with phosphoric acid to increase its ability to adsorb the oxygen [23]. Bai et al. fabricated phosphate-modified gC3N4 via acid-hydrothermal post-treatment for enhanced H2O2 production [45].
On the other hand, metal doping is also an effective method to promote the photocatalytic activity of g-C3N4. Wang et al. reported that Fe-doped g-C3N4 possessed better photocatalytic benzene oxidation performance [32]. Li et al. created a vertical channel via intercalating alkalis into the interlayer space in g-C3N4 to effectively quench charge recombination and promote electron delivery [46]. However, to our best knowledge, few literatures on the photocatalytic performance of anions/metal co-functionalized g-C3N4 have been reported. Also, the conversion pathway of photocatalytic NO oxidation as a key issue has not been clearly revealed.
Inspired by the introduction of metal ions between the gC3N4 layers and the modification of g-C3N4 surface by the inorganic nonmetal redox-inert anions, so we propose that simultaneous modification between layers and surfaces of CN could significantly enhanced the NO purification performance. Here, the PO4/K co-functionalized g-C3N4 (labeled as PO4-CN-K) was synthesized via a facile co-pyrolysis of thiourea mixed with K3PO4. The experimental and theoretical methods are highly combined to reveal the promotion mechanisms of NO conversion in terms of enhanced reactants activation, charge transfer and photocatalytic efficiency. The present work could provide a novel strategy for enhancing the photocatalytic efficiency and represent new insights into g-C3N4 photocatalysis mechanism for air purification.
Herein, 10.0 g thiourea and potassium phosphate (K3PO4) with different mass (0.110, 0.018, 0.036, 0.073 g) were dissolved in 20 mL water in four alumina crucibles (50 mL), respectively. Then the obtained solution was dried in 60 ℃ overnight to get the solid precursors, which were then placed into a muffle furnace, and heated to 550 ℃ at a heating rate of 15 ℃/min for 2 h. After the thermal treatment, the crucible was cooled down to room temperature in the muffle furnace. The obtained samples were collected and labelled as PO4-CN-K-0.11, PO4-CN-K-0.01, PO4-CNK-0.03 and PO4-CN-K-0.07. For comparison, the pristine CN was prepared without adding K3PO4 and labelled as CN. Additionally, as a reference, 10 g thiourea and 0.12 g potassium bromide (with the same potassium atomic content as the most active sample) were prepared by the same method to obtain k-doped CN (CN-K).
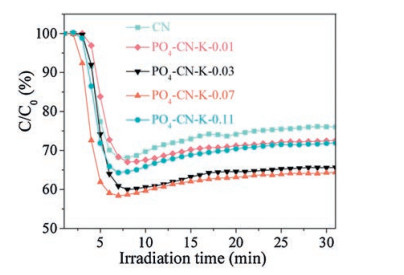
The photocatalytic NO purification performance of CN and PO4-CN-K catalysts under visible light irradiation is evaluated by measuring the concentration of NO with NOx analyzer. As can be seen in Fig. 1, the photocatalytic NO removal ratio of pure CN is 22.7%. When potassium phosphate is intercalated into CN, the photocatalytic activity is obviously improved. As a reference, the photocatalytic NO removal ratio of K-doped g-C3N4 (CN-K) is 26.2% (Fig. S1 in Supporting information). When the mass ratio of potassium phosphate and thiourea is controlled at 0.73%, the NO removal ratio of PO4-CN-K-0.07 reaches as high as 35.2%. Therefore, the result indicates that the PO4/K co-functionalization is novel and feasible strategy to enhance the photocatalytic activity of CN.

|
Download:
|
| Fig. 1. Photocatalytic activity for NO purification for all the samples. | |
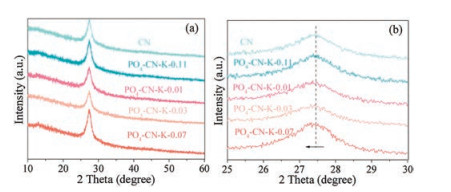
The X-ray diffraction (XRD) patterns of the samples reveal two characteristic diffraction peaks (13.1° and 27.4°) associated respectively with the in-plane repeated unites and the periodic graphitic stacking of the conjugate aromatic system (Fig. 2). The XRD pattern of PO4-CN-K is similar to that of pure CN, indicating the retained CN frameworks with K3PO4 incorporation [18]. It is worth noting that the characteristic peak corresponding to the dominant (002) peak is shifted toward lower angle due to the intercalation of potassium phosphate.

|
Download:
|
| Fig. 2. XRD of CN and the PO4/K co-doping CN samples with different K3PO4 content (a) and the enlarged profile of the (002) diffraction region (b). | |
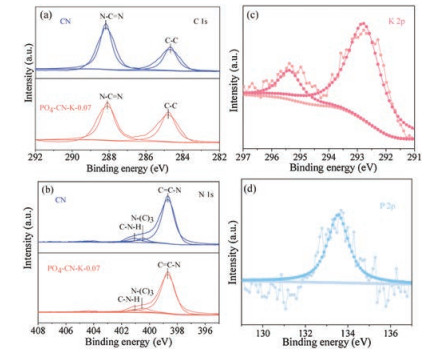
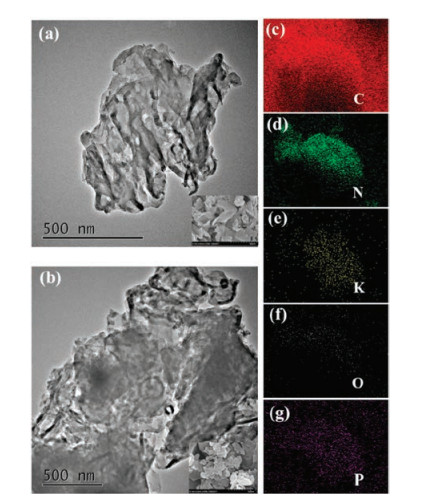
Subsequently, the X-ray photoelectron spectroscopy (XPS) was used to further investigate the composition and chemical state of the samples (Fig. 3, Table S1 in Supporting information). The high-resolution XPS spectra of C 1s (Fig. 3a) exhibits two main peaks arising from the sp2-bonded carbon in the N-containing aromatic rings (C–N = C) at 288.2 eV and the sp2 C─C bonds at 284.8 eV, respectively. As shown in Fig. 3b, the N 1s spectra can be deconvoluted into three peaks at 398.6 eV, 400.5 eV and 401.1 eV, corresponding to the sp2-bonded N involved in the triazine rings (C–N = C), the tertiary nitrogen N-(C)3 groups, and the amino functions (C-N-H), respectively [47, 48]. The binding energies of K 2p3/2 and K 2p1/2 peaks in PO4-CN-K-0.07 are located at 292.78 eV and 295.38 eV, which demonstrates that the K+ has been successfully doped into CN [49]. A binding energy of P 2p is located at around 133.5 eV, implying that the phosphorus is in pentavalent-oxidation state (P5+) and exists in the form of P─O bond [50]. The SEM and TEM images (Figs. 4a and b ) display that PO4-CN-K-0.07 possesses a typical layered structure, which is similar than that of pristine CN. Additionally, EDX elemental mapping of PO4-CN-K-0.07 shows that the elements C, N, K, O, and P are distributed uniformly across the whole samples (Figs. 4c-g).

|
Download:
|
| Fig. 3. XPS spectra of CN and PO4-CN-K-0.07, (a) C 1s, (b) N 1s, (c) K 2p, (d) P 2p. | |

|
Download:
|
| Fig. 4. SEM and TEM images of CN (a) and PO4-CN-K-0.07 (b), FESEM-EDX elemental mapping of PO4-CN-K-0.07 (c–g). | |
From the above characterization results, it can be concluded that the K3PO4 has been successfully doped into CN. Next, the close combination of experimental and theoretical calculation is used to explore the essential reasons for the enhanced photocatalytic activity. First, we construct the PO4/K co-doped could not be accommodated between the CN layers, so the phosphate group is inclined to attach on the CN surface (Figs. 5a and b). The adsorption and activation capacities of O2, NO, and H2O on both CN and PO4-CN-K-0.07 are calculated using DFT. calculations. As shown in Fig. 5c, more efficient charge transfer is observed between O2 and PO4-CN-K-0.07 than that between O2 and CN. The adsorption energy (Eads) for the O2 molecule is increased from -0.19 eV on CN to -0.42 eV on PO4-CN-K-0.07, resulting in the O2 accepting electrons from 0.14 e (CN) to 0.23 e (PO4-CN-K-0.07). The NO adsorption capacity on PO4-CN-K-0.07 is also increased from -0.1 eV to -0.12 eV. The NO loses more electrons from -0.03 e on CN to -0.07 e on PO4-CN-K-0.07, thus enhancing the activation capacity of NO over PO4-CN-K-0.07. Additionally, the water adsorption capacity on PO4-CN-K-0.07 is also increased significantly from -0.17 eV to -0.51 eV, validating that the activation capacity of water molecules is enhanced to generate more hydroxyl radicals to oxide the pollutant. The specific calculation parameters are listed in Table S2 in Supporting information.

|
Download:
|
| Fig. 5. Optimized local structures of K3PO4 modified CN at surface (a) and intercalated sites (b). DMPO ESR spectra in methanol dispersion for O2- (d) and aqueous dispersion for OH (e) are carried out in both dark and visible light irradiation for 15 min, respectively. (c) Charge difference density distribution of optimized O2, NO, H2O, adsorption in CN and PO4-CN-K-0.07, respectively, the charge accumulation and depletion are in blue and yellow, respectively; pink, purple, rose red, gray, and red spheres represent C, N, K, P, and O atoms respectively; E0 stands for the total energy, negative means heat release. | |
It is well known that reactive oxygen species (ROS), such as superoxide (·O2-), hydroxyl (·OH), as highly active and green oxidants, are of great significance for environmental chemistry and oxidation [51]. The standard 5, 5-dimethyl-pyrroline N-oxidize (DMPO) spin-trapping electron spin resonance (ESR) method is used to investigate the generation of active radicals on CN and PO4-CN-K-0.07. As can been seen in Figs. 5d and e, much stronger DMPO-·O2- and DMPO-·OH signals are detected on PO4-CN-K-0.07 than that on pure CN. The PO4-CN-K could improve adsorption and activation of O2 and H2O, and expedite spatial charge separation, which contribute directly to produce more superoxide and hydroxyl radicals, thus enhancing the purification ability of NO.
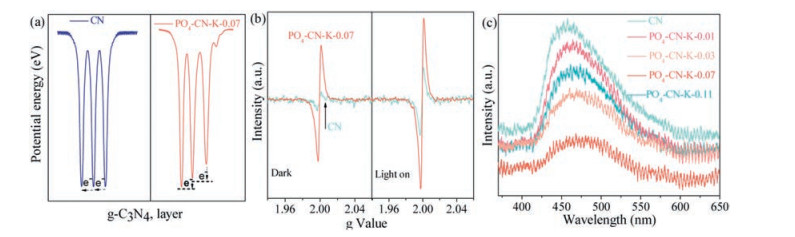
Next, we further exploreed the photo-generated electrons/holes separation process affecting the production of active substances. As shown in Fig. 6a, the potential energies of the first layer and the second layer are obviously increased after introducing PO4- and K+ compared with the pure CN, implying that the PO4/ K co-functionalization could reduce the interlayer energy barrier and promote the electron transfer between the adjacent layers. Simultaneously, the stronger EPR signal intensity of PO4-CN-K-0.07 is observed, suggesting that a more efficient electronic mobility and electronic transport efficiency (Fig. 6b). In addition, the PL peak quenching also indicates that the carrier recombination rate of PO4-CN-K is significantly reduced (Fig. 6c). Therefore, the PO4/K co-functionalization could promote the charge separation and inhibits photo-generated carrier recombination, thus accelerating the production of ROS to improve the photocatalytic NO purification efficiency.

|
Download:
|
| Fig. 6. Calculated electrostatic potential of CN and PO4-CN-K-0.07 (a), room temperature solid state EPR spectra of CN and PO4-CN-K-0.07 (b), PL spectra of the as-prepared samples (c). | |
The conversion process of NO adsorption and reaction in time sequence can be dynamically monitored by in situ FT-IR. Firstly, the background spectra are recorded before NO is injected into the reaction chamber. A series of characteristic peaks will appear immediately after passing the mixture of NO and O2 at 25 ℃ under dark condition (Fig. 7a). The NO2 (951 cm-1) adsorption band is observed on the surface of CN [52]. Some of these NO2 turns into chelated nitrites (868 cm-1), NO2- (847 cm-1), monodentate nitrites (1024 cm-1), bi/monodentate nitrite (1106 cm-1), which can be attributed to the activation of NO by reactive ·O2- species produced by the lone pair electrons given to O2 by pyridine N atoms [52-56]. In the case of PO4-CN-K-0.07 (Fig. 7b), the adsorption peaks in Fig. 7a appear as well. Additionally, there are many new characteristic peaks. The new bands at 1005 cm-1 can be assigned to the stretching vibration of nitrates [55]. Besides, NO-/NOH at 1162 cm-1, NO2 at 2060 cm-1, and N2O4 at 2170 cm-1 are adsorbed on the surface of PO4-CN-K-0.07 [57-59]. It is worth nothing that after the doping of PO43- and K+ into CN, an interesting peak appears at 2133 cm-1, which belongs to NO+ (Table 1). The transformation of this intermediate NO+ makes it easier for NO to be oxidized into the final product [60]. These final products accumulated on the photocatalyst surface can be removed easily by water washing and the photocatalyst can be regenerated via this facile method.

|
Download:
|
| Fig. 7. In situ FT-IR spectra of NO adsorption and photocatalytic reaction processes over CN and PO4-CN-K-0.07 under visible light irradiation. | |
|
|
Table 1 Assignments of the FT-IR bands observed during the adsorption-reaction of NO on CN and PO4-CN-K-0.07 followed by visible irradiation. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Once the adsorption equilibrium is achieved, the timedependent IR spectra of CN and PO4-CN-K-0.07 are monitored dynamically under visible-light irradiation. As shown in the photocatalytic NO oxidation process over CN (Fig. 7c), the bands at 1031 cm-1 and 982 cm-1 can be assigned as NO3-, which is a kind of target product and gradually accumulates on the surface of catalyst [61]. Under light-illumination, there is no new adsorption bands on the surface of PO4-CN-K-0.07 compared with under dark conditions, but the peak intensity of 866 and 1102 cm-1 as the final product is significantly enhanced [52, 55]. Notably, the adsorption intensity of final products is much stronger on PO4-CN-K in comparison with those on the pure CN, which suggests more efficient transformation of NO on PO4-CN-K. Based on above results, the reaction mechanisms of photocatalytic NO oxidization by PO4-CN-K-0.07 are proposed as following two channel reactions.
Reaction pathway 1:

|
Reaction pathway 2:

|
In summary, the PO4/K co-functionalized carbon nitride was synthesized via a facile co-pyrolysis of thiourea mixed with K3PO4 for the first time. The unique electronic structure of PO4-CN-K was revealed via a combined experimental and theoretical approach. The surface modification with phosphate could enhance the adsorption of reactants and interlayer incorporation with potassium could reduce the interlayer potential barrier and promote charge separation, which in all contributed to an enhancement in photocatalytic NO purification under visible light irradiation. The corresponding conversion pathway of NO on the photocatalyst surface was disclosed via time-dependent in situ FT-IR. The mechanism of two channel reactions for photocatalytic NO oxidization was proposed. This work could provide a new perspective for the design of efficient photocatalyst for air purification.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21822601, 21777011 and 21501016), the Innovative Research Team of Chongqing (No. CXTDG201602014), the Natural Science Foundation of Chongqing (No. cstc2017jcyjBX0052), and the Plan for "National Youth Talents" of the Organization Department of the Central Committee. The authors also acknowledge AM-HPC in Suzhou, China for computational support.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.03.016
| [1] |
I.F. Teixeira, E.C.M. Barbosa, S.C.E. Tsang, et al., Chem. Soc. Rev. 47 (2018) 7783-7817. DOI:10.1039/C8CS00479J |
| [2] |
X. Li, W. Zhang, W. Cui, et al., Appl. Catal. B:Environ. 221 (2018) 482-489. DOI:10.1016/j.apcatb.2017.09.046 |
| [3] |
J.D. Li, X.L. Zhang, F. Raziq, et al., Appl. Catal. B:Environ. 218 (2017) 60-67. DOI:10.1016/j.apcatb.2017.06.038 |
| [4] |
Z.Y. Wang, Y. Huang, W.K. Ho, et al., Appl. Catal. B:Environ. 199 (2016) 123-133. DOI:10.1016/j.apcatb.2016.06.027 |
| [5] |
F. Chen, H.W. Huang, L. Guo, et al., Angew. Chem. Int. Ed. (2019). DOI:10.1002/anie.201901361 |
| [6] |
Y. Huang, Y.L. Liang, Y.F. Rao, et al., Environ. Sci. Technol. 51 (2017) 2924-2933. DOI:10.1021/acs.est.6b04460 |
| [7] |
C. Buck, N. Skillen, J. Robertson, et al., Chin. Chem. Lett. 29 (2018) 773-777. DOI:10.1016/j.cclet.2018.04.022 |
| [8] |
Y. Wang, C.X. Feng, M. Zhang, et al., Appl. Catal. B:Environ. 104 (2011) 268-274. DOI:10.1016/j.apcatb.2011.03.020 |
| [9] |
D. Zhao, C.C. Chen, C.L. Yu, et al., J. Phys. Chem. C 113 (2009) 13160-13165. DOI:10.1021/jp9002774 |
| [10] |
H. Wang, W. He, X. Dong, et al., Sci. Bull. 63 (2018) 117-125. DOI:10.1016/j.scib.2017.12.013 |
| [11] |
E.S. Kwak, W. Lee, N.G. Park, et al., Adv. Funct. Mater. 19 (2009) 1093-1099. DOI:10.1002/adfm.v19:7 |
| [12] |
P.W. Huo, J.Z. Li, Z.F. Ye, et al., Chin. Chem. Lett. 28 (2017) 2259-2262. DOI:10.1016/j.cclet.2017.09.067 |
| [13] |
B.X. Zhao, P.Y. Zhang, Catal. Commun. 10 (2009) 1184-1187. DOI:10.1016/j.catcom.2009.01.017 |
| [14] |
H.W. Huang, X.W. Li, J.J. Wang, et al., ACS Catal. 5 (2015) 4094-4103. DOI:10.1021/acscatal.5b00444 |
| [15] |
E. Kroke, M. Schwarz, Coord. Chem. Rev. 248 (2004) 493-532. DOI:10.1016/j.ccr.2004.02.001 |
| [16] |
Y.J. Zhang, M. Antonietti, Chem. Asian J. 5 (2010) 1307-1311. |
| [17] |
P. Chen, Y. Sun, H. Liu, et al., Nanoscale 11 (2019) 2366-2373. DOI:10.1039/C8NR09147A |
| [18] |
X.C. Wang, K. Maeda, A. Thomas, et al., Nat. Mater. 8 (2009) 76-80. DOI:10.1038/nmat2317 |
| [19] |
S.C. Yan, Z.S. Li, Z.G. Zou, Langmuir 25 (2009) 10397-10401. DOI:10.1021/la900923z |
| [20] |
G. Liu, P. Niu, C.H. Sun, et al., J. Am. Chem. Soc. 132 (2010) 11642-11648. DOI:10.1021/ja103798k |
| [21] |
S.W. Cao, J.X. Low, J.G. Yu, et al., Adv. Mater. 27 (2015) 2150-2176. DOI:10.1002/adma.201500033 |
| [22] |
J. Li, X. Dong, Y. Sun, et al., Appl. Catal. B:Environ. 239 (2018) 187-195. DOI:10.1016/j.apcatb.2018.08.019 |
| [23] |
C. Liu, L.Q. Jing, L.M. He, et al., Chem. Commun. 50 (2014) 1999-2001. DOI:10.1039/c3cc47398h |
| [24] |
Y.L. Chen, Z.S. Zhan, J.H. Wang, et al., Chin. Chem. Lett. 29 (2018) 437-440. DOI:10.1016/j.cclet.2017.08.028 |
| [25] |
Y. Zheng, L.H. Lin, B. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 12868-12884. DOI:10.1002/anie.v54.44 |
| [26] |
H.F. Shi, G.Q. Chen, C.L. Zhang, et al., ACS Catal. 4 (2014) 3637-3643. DOI:10.1021/cs500848f |
| [27] |
Z.Z. Lin, X.C. Wang, Angew. Chem. Int. Ed. 52 (2013) 1735-1738. DOI:10.1002/anie.v52.6 |
| [28] |
J.S. Zhang, M.W. Zhang, C. Yang, et al., Adv. Mater. 26 (2014) 4121-4126. DOI:10.1002/adma.v26.24 |
| [29] |
J. Li, Z. Zhang, W. Cui, et al., ACS Catal. 8 (2018) 8376-8385. DOI:10.1021/acscatal.8b02459 |
| [30] |
Z. Xu, S.S. Xu, N. Li, et al., ACS Sustain. Chem. Eng. 5 (2017) 9667-9672. DOI:10.1021/acssuschemeng.7b03088 |
| [31] |
C. Chang, Y. Fu, M. Hu, et al., Appl. Catal. B:Environ. 142 (2013) 553-560. |
| [32] |
X.F. Chen, J.S. Zhang, X.Z. Fu, et al., J. Am. Chem. Soc. 131 (2009) 11658-11659. DOI:10.1021/ja903923s |
| [33] |
W. Cui, J. Li, Y. Sun, et al., Appl. Catal. B:Environ. 237 (2018) 938-946. DOI:10.1016/j.apcatb.2018.06.071 |
| [34] |
Y.J. Zhou, L.X. Zhang, J.J. Liu, et al., J. Mater. Chem. A 3 (2015) 3862-3867. DOI:10.1039/C4TA05292G |
| [35] |
Z.Y. Wang, M.J. Chen, Y. Huang, et al., Appl. Catal. B:Environ. 239 (2018) 352-361. DOI:10.1016/j.apcatb.2018.08.030 |
| [36] |
J.L. Zhang, Y.M. Wu, M.Y. Xing, et al., Energy Environ. Sci. 3 (2010) 715-726. DOI:10.1039/b927575d |
| [37] |
H.J. Yu, J.Y. Li, Y.H. Zhang, et al., Angew. Chem. Int. Ed. (2018). DOI:10.1002/anie.201813967 |
| [38] |
L.Q. Jing, J. Zhou, J.R. Durrant, et al., Energy Environ. Sci. 5 (2012) 6552-6558. DOI:10.1039/c2ee03383f |
| [39] |
Y. Cao, L.Q. Jing, X. Shi, et al., Phys. Chem. Chem. Phys. 14 (2012) 8530-8536. DOI:10.1039/c2cp41167a |
| [40] |
L. Lin, W. Lin, J.L. Xie, et al., Appl. Catal. B:Environ. 75 (2007) 52-58. DOI:10.1016/j.apcatb.2007.03.016 |
| [41] |
P. Mohapatra, K.M. Parida, J. Mol. Catal. A-Chem. 258 (2006) 118-123. DOI:10.1016/j.molcata.2006.05.002 |
| [42] |
S.K. Samantaray, P. Mohapatra, K. Parida, J. Mol. Catal. A-Chem. 198 (2003) 277-287. DOI:10.1016/S1381-1169(02)00693-3 |
| [43] |
H. Park, W. Choi, J. Phys. Chem. B 108 (2004) 4086-4093. DOI:10.1021/jp036735i |
| [44] |
M.S. Vohra, S. Kim, W. Choi, J. Photochem. Photobiol. A 160 (2003) 55-60. DOI:10.1016/S1010-6030(03)00221-1 |
| [45] |
J. Bai, Y.Z. Sun, M.Y. Li, et al., Relat. Mater. 87 (2018) 1-9. DOI:10.1016/j.diamond.2018.05.004 |
| [46] |
J.Y. Li, W. Cui, Y.J. Sun, et al., J. Mater. Chem. A 5 (2017) 9358-9364. DOI:10.1039/C7TA02183F |
| [47] |
A. Thomas, A. Fischer, F. Goettmann, et al., J. Mater. Chem. 18 (2008) 4893-4908. DOI:10.1039/b800274f |
| [48] |
G.G. Zhang, J.S. Zhang, M.W. Zhang, et al., J. Mater. Chem. 22 (2012) 8083-8091. DOI:10.1039/c2jm00097k |
| [49] |
W. Cui, J.Y. Li, W.L. Cen, et al., J. Catal. 352 (2017) 351-360. DOI:10.1016/j.jcat.2017.05.017 |
| [50] |
D. Zhao, C.C. Chen, Y.F. Wang, et al., J. Phys. Chem. C 112 (2008) 5993-6001. DOI:10.1021/jp712049c |
| [51] |
H.W. Huang, S.C. Tu, C. Zeng, et al., Angew. Chem. Int. Ed. 56 (2017) 11860-11864. DOI:10.1002/anie.201706549 |
| [52] |
K.I. Hadjiivanov, Catal. Rev. 42 (2000) 71-144. DOI:10.1081/CR-100100260 |
| [53] |
W. He, Y. Sun, G. Jiang, et al., Appl. Catal. B:Environ. 232 (2018) 340-347. DOI:10.1016/j.apcatb.2018.03.047 |
| [54] |
J.C.S. Wu, Y.T. Cheng, J. Catal. 237 (2006) 393-404. DOI:10.1016/j.jcat.2005.11.023 |
| [55] |
K. Hadjiivanov, V. Avreyska, D. Klissurski, et al., Langmuir 18 (2012) 1619-1625. |
| [56] |
Y. Zhou, Z.Y. Zhao, F. Wang, et al., J. Hazard. Mater. 307 (2016) 163-172. DOI:10.1016/j.jhazmat.2015.12.072 |
| [57] |
M. Kantcheva, J. Catal. 204 (2001) 479-494. DOI:10.1006/jcat.2001.3413 |
| [58] |
H. Miyata, S. Konishi, T. Ohno, et al., J. Chem. Soc. Faraday T. 91 (1995) 1557-1562. DOI:10.1039/ft9959101557 |
| [59] |
J.L. Hardwick, J.C.D. Brand, Can. J. Phys. 54 (1976) 80-91. DOI:10.1139/p76-010 |
| [60] |
R.F.W. Bader, P.L.A. Popelier, T.A. Keith, Angew. Chem. Int. Ed. 33 (1994) 620-631. |
| [61] |
W. He, Y. Sun, G. Jiang, et al., Appl. Catal. B:Environ. 239 (2018) 619-627. DOI:10.1016/j.apcatb.2018.08.064 |