 2019, Vol. 30
2019, Vol. 30 


b University of Chinese Academy of Sciences, Beijing 100049, China;
c Ningbo Urban Environment Observation and Research Station-NUEORS, Institute of Urban Environment, Chinese Academy of Sciences, Ningbo 315800, China;
d State Key Joint Laboratory of Environment Simulation and Pollution Control, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
Iron-based zeolites (mostly Fe-ZSM-5 and Fe-Beta) have been extensively investigated because of their high activity and thermal stability for the selective catalytic reduction of NOx with NH3 (NH3-SCR) [1-6]. Compared with ZSM-5, Beta zeolite is an attractive host for iron and Fe-Beta zeolite has higher activity than Fe-ZSM-5 in some catalytic reactions [7, 8]. Many studies focused on Fe-Beta catalysts for N2O decomposition [8-11] and NH3-SCR, especially on the active sites and hydrothermal stability [1, 12, 13]. Previous studies suggested that the low-temperature NH3-SCR activity of Fe-Beta could be enhanced by bimetallic doping (such as Cu [14], Mn [15], Ce [16, 17]) and high-temperature H2-pretreatment [2, 6].
Hierarchical zeolites contain both micro and meso-/macrospores, which can eliminate diffusion limitations and promote the accessibility of reactants to the active sites [18-21]. Thepredominant approach for the synthesis of hierarchical structure zeolites is demetallation (desiliconization and dealumination) and recrystallization with the surfactants [19, 22]. Previous studies suggested that NH3-SCR activity was enhanced by introducing hierarchical structure to Fe-ZSM-5 [23, 24]. However, there are few open literatures about hierarchical Fe-Beta for NH3-SCR.
In this work, hierarchical Fe-Beta zeolites catalysts were prepared by hydrothermal synthesis and evaluated for NH3-SCR. The preparation procedure and activity tests are shown in the Support information. It was found that the low-temperature activity of hierarchical Fe-Beta was higher than that of Fe-Beta. The structure and activity relationship was discussed in detail.
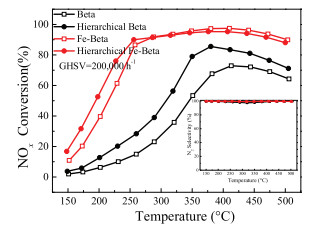
As shown in Fig. 1, it was clear that the Betawas active for the SCR reaction in a narrow temperature range of 375–500 ℃. Acidity may account for its catalytic performance [25, 26]. Hierarchical Beta zeolite showed obviously improved NO conversions in the whole temperature range. This result indicated that hierarchical Beta zeolite may have more acid sites than Beta, which will be verified in the subsequent NH3-TPD and NH3 adsorption sections. After the doping of Fe, the Fe-Beta and hierarchical Fe-Beta indicated highly improved SCR activity as compared with Beta and hierarchical Beta in the whole temperature of 150–500 ℃. Hierarchical Fe-Beta showed enhanced low-temperature SCR activity compared with Fe-Beta obtained using the same ion-exchanged procedure. All of these catalysts presented excellent N2 selectivity. The relationship of structure and activity was discussed in detail as follows.

|
Download:
|
| Fig. 1. NOx conversion and N2 selectivity (inserted) results of Fe-Beta and Beta catalysts. Reaction conditions: [NO] = [NH3] =500 ppm, [O2] =5 vol%, N2 balance and GHSV = 200, 000 h-1. | |
{kind=link}
The XRD patterns (Fig. S1 in Supporting information) show that all samples are phase-pure zeolites with the BEA framework structure. The peak intensity of Fe-Beta and hierarchical Fe-Beta declined, which may be due to the collapse or blocking of the partial microspore of zeolite. FeOx species were not detected, which may be due to the Fe species existing as amorphous or high dispersion on the zeolite support [27, 28].
The textural properties in Table S1 and Fig. S2 (Supporting information) show that the total volume, external surface area and BET surface area of hierarchical Beta are larger than those of Beta, indicating that mesopores and/or macropores are introduced into Beta to form hierarchical structure [19, 20]. After doping with Fe, the BET surface area of both Fe-Beta and hierarchical Fe-Beta decreased, indicating that the partial microspore may be blocked, in agreement with the XRD results. ICP analysis revealed that the Fe content of Fe-Beta and hierarchical Fe-Beta was 2.82% and 3.71%, respectively, suggesting that hierarchical Fe-Beta could achieve a large amount of iron at the exchange sites under the identical ionexchange procedure.
Comparing TEM images of hierarchical Fe-Beta (Figs. S3c and d in Supporting information) with those of Fe-Beta (Figs. S3a and b in Supporting information), the smaller crystal size and some hollow Beta structure were obtained during the hydrothermal synthesis, indicating the formation of micro-macroporous structure.This result is in accordance with the BET results. In Fig. S3c, the hollow zeolite was made of a polycrystalline micro-mesoporous shell and macroporecore.Previous studies suggested that hollow Beta single crystals were much more difficult to prepare [18]. The absence of Al zoning prohibited the use of the dissolution-recrystallization strategy employing in the synthesis of hollow Beta zeolite single crystals [29].
UV-vis DR spectroscopy is an effective and common method for determining the nature and distribution of Fe species in Fe-zeolites. Fig. S4a (Supporting information) shows the UV–vis DR spectra of the Fe-Betasamples.The spectra were deconvoluted into sub-sands, which can be attributed to specific Fe species in the Fe-zeolites. The bands at 211 and 277 nm were respectively assigned to isolated Fe3+ ions in tetrahedral and octahedral coordination environments [30].Thebands at 300–400 nm indicated the presence of small oligomers Fex3+Oy in the zeolite channels [8, 28], and the adsorption bands above 400 nm were attributed to the presence of bulky Fe2O3 nanoparticles on the external surface of the zeolite [31, 32]. Fig. S4b (Supporting information) shows the UV–vis DR spectra of hierarchical Fe-Beta samples, the peak attributions are similar to Fe-Beta samples. The amount of Fe species was estimated semi-quantitatively based on the relative intensities of these sub-bands derived from UV–vis DR spectra and the total Fe loading in the zeolite. The data inTable S2 in Supporting information shows that isolated Fe3+ ions are the predominant species in Fe-Beta and hierarchical Fe-Beta catalysts. Previous studies suggested isolated Fe3+ species are active sites for NH3-SCR on Fe-Beta catalysts [4, 5, 25, 33, 34]. When the Fe loadings were considered, the hierarchical Fe-Beta had the larger content of isolated Fe3+ species than Fe-Beta, resulted in the enhanced NH3-SCR performance.
EPR spectroscopy was further employed to identify the nature of different Fe species, the corresponding results are illustrated in Fig. S5 (Supporting information). Signals at g = 2.0, g = 4.3 and g = 8.8 were observed for Fe-Beta and hierarchical Fe-Beta. The signals at g = 4.3, g = 8.8 and g = 2 were generally attributed to isolated Fe species in tetrahedral and distorted tetrahedral coordination [30, 35], isolated Fe species with a distorted octahedral environment [8, 13] and isolated Fe species in a high symmetry octahedral coordination or oligonuclear FexOy clusters [8, 28], respectively. Overall, the EPR signal intensity of hierarchical Fe-Beta was higher than Fe-Beta, illustrating the amount of isolated Fe species on hierarchical Fe-Beta was higher than that of Fe-Beta, in agreement with UV–vis DRS results.
In order to investigate the redox performance of the samples, the H2-TPR experiments were conducted and the results are shown in Fig. 2. There is no clear H2 consumption signal for the Beta and hierarchical Beta. Two reduction peaks appeared on the Fe-Beta and hierarchical Fe-Beta. The overlapped reduction peaks centered at 354 ℃ (369 ℃) and 441 ℃ (499 ℃) were attributed tothe reduction of isolated Fe3+ to Fe2+ [36] and oligomeric iron oxo species or tiny Fe2O3 into Fe3O4, respectively [37-39]. The reduction temperature of Fe species over hierarchical Fe-Beta was lower than Fe-Beta, which may be due to the better dispersion of Fe species. The amount of H2 consumption of hierarchical Fe-Beta was larger than Fe-Beta, indicating that hierarchical Fe-Beta catalyst had more Fe active sites. The better redox performance resulted in the enhanced NH3-SCR activity on the hierarchical Fe-Beta.

|
Download:
|
| Fig. 2. H2-TPR results of the different Beta samples. | |
{kind=link}
In view of the analysis of UV–vis DRS, EPR and H2-TPR results above, we have confirmed that more Fe active species existed on the hierarchical Fe-Beta and dispersed better than those on FeBeta, which may be due to the specific hierarchical structure.
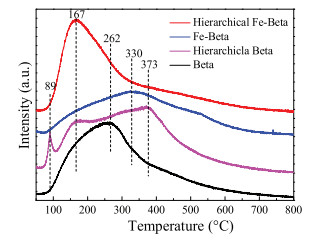
To identify the strength of the acid sites of hierarchical Fe-Beta, the NH3-TPD experiments were carried out and the results are shown in Fig. 3. Beta zeolite showed NH3 desorption peaks at the range of 100–500 ℃, assigning to the desorption of ammonia species from weak Lewis acid sites (167 ℃), strong Lewis acid sites (262 ℃ and 330 ℃) and strong Brønsted acid sites (373 ℃) [25, 37, 40]. The intensity of the desorption peak at 262 ℃ was much stronger than that of peak at 373 ℃, suggesting that Beta zeolite had more Lewis acid sites because of its structural defects caused by the unsaturated bonding of silicon [41, 42]. When forming the hierarchical structure, the peaks shifted to the hightemperature, demonstrating the formation of the stronger acid sites. The peak at 89 ℃ on hierarchical Beta may be attributed to physisorbed NH3 [39, 43]. The number of acid sites of hierarchical Beta also increased, which resulted in the enhanced SCR activity, compared with Beta zeolite (Fig. 1). When Fe was introduced into Beta, the NH3 storage capacity decreased, and the desorption peak shifted to high-temperature. However, when Fe was exchanged to hierarchical Beta, the NH3 desorption peak shifted to the lowtemperature, to form more weak Lewis acid sites (167 ℃), which is beneficial to the adsorption and activation of NH3.

|
Download:
|
| Fig. 3. NH3-TPD profiles of different Beta zeolites. | |
{kind=link}
In order to understand the adsorption performance and reaction mechanism, in situ DRIFTS experiments were carried out for Fe-Beta and hierarchical Fe-Beta (Fig. S6 in Supporting information). After the adsorption of NH3 and N2 purge, the NH3adsorbed peaks appeared on both of the catalysts.Then egative bands at 3773, 3721, 3604 cm-1 was assigned to Al-OH groups, terminal Si-OH, isolated bridging acidic hydroxyl stretching vibrations (Si(OH)Al), respectively [44-46]. The negative band at 3675 cm-1 only appeared on hierarchical Fe-Beta, which was attributed to hydroxyls bonded on extra-framework Al species [47], due to hierarchical structure. This result indicated that hierarchical structure introduced more exchanged sites and acid sites, which led to the better SCR activity. In the N-H strecthing regions (3100-3500 cm-1), the bands corresponding to the asymmetric strecthing (3343 cm-1) and symmetric strecthing (3273 and 3192 cm-1) of coordinated NH3 on Lewis acid sites were observed, respectively [48]. The bands at 1478 cm-1 and 1163 cm-1 were attributed to the NH4+ ions adsorbed on the Brønsted acid sites and coordinated NH3 adsorbed on the Lewis acid sites, respectively [48-50]. The band at 896 cm-1 could be assigned to T-O-T framework vibrations by Fe species [7]. The peak intensity at 896 cm-1 of hierarchical Fe-Betawas higherthan that of Fe-Beta, illustrating more Fe was incorporated into hierarchical Fe-Beta, in agreement with the ICP, EPR and H2-TPR results. Overall, the intensity of NH3 adsorption peaks for hierarchical Fe-Beta was higher than those for Fe-Beta, suggesting more acid sites existing on hierarchical Fe-Beta than those on Fe-Beta, in accordance with NH3-TPD results.
The NO adsorbed spectra were shown in Fig. S6b. No peak for NOx adsorbed species was clearly observed, suggesting that Fe-Beta and hierarchical Fe-Beta did not adsorb NOx species. The NOx adsorbed species were not detected on the Fe-Beta and hierarchical Fe-Beta, but the excellent NOx conversion could be obtained, illustrating that the NH3-SCR reaction may mainly carried out through Eley-Rideal (E-R) pathway with gaseous NO reacting with adsorbed NH3 species on both of the catalysts.
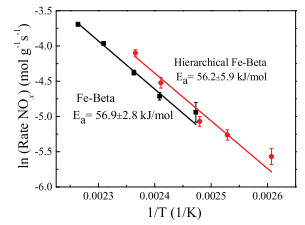
In order to obtain further understanding of the intrinsic effect of hierarchical structure on the NH3-SCR reaction, the kinetic experiments were performed on the Fe-Beta and hierarchical Fe-Beta, as shown in Fig. 4. The apparent activation energy obtained from the Arrhenius plots for Fe-Beta and hierarchical Fe-Beta was 56.9 and 56.2 kJ/mol, respectively, in agreement with those for Fe-zeolite reported in the previous literature (Ea=40~54 kJ/mol) [31, 51, 52]. The two catalysts had similar activation energy, indicating an identical rate controlling mechanism [13]. Since there were no mass transfer related limitations, hierarchical Fe-Beta catalyst had better SCRactivity, demonstrating the higher activity was mainly causedby higher amount of Fe active sites [53], in accordance with the H2-TPR and UV–vis DRS results.

|
Download:
|
| Fig. 4. Arrhenius plots of the SCR reaction rates over Fe-Beta and hierarchical FeBeta in the feed gas of 500 ppm NO/ 500 ppm NH3/ 5.0% vol% O2/ N2 (500 mL/min, 40 mg catalysts, GHSV: 400, 000 h-1). | |
{kind=link}
In summary, hierarchical Beta was prepared by hydrothermal synthesis. The BET and TEM results confirmed that the meso/ macrospores were introduced to Beta zeolite. Hierarchical Fe-Beta exhibited better low-temperature activity than Fe-Beta. The results of UV–vis DRS, EPR and H2-TPR indicated that isolated Fe3+, oligomers and Fe2O3 coexisted on Fe-Beta and hierarchical Fe-Beta. The enhanced activity of the hierarchical Fe-Beta was related to the prominent presence of highly dispersed Fe active species, which may be because that the hierarchical structure introduced more defects to provide more ion-exchange sites for Fe species. The kinetic experiments demonstrated that the activate energy of FeBeta and hierarchical Fe-Beta was 56.2 and 56.9 kJ/mol, respectively, suggesting an identical rate controlling mechanism. The NH3-TPD and DRIFTS results of NH3 adsorption indicated that the hierarchical Fe-Beta had more acid sites for the adsorption and activation of NH3. The adsorption of NOx was not clearly observed on Fe-Beta or hierarchical Fe-Beta, indicating that NH3-SCR process of both of the two catalysts mainly followed the Eley-Rideal (E-R) reaction mechanism.
AcknowledgmentsWe gratefully acknowledge the financial supports from the National Natural Science Foundation of China (Nos. 51822811, 21637005), the National Key R&D Program of China (Nos. 2017YFC0212502, 2017YFC0211101), and the Young Talent Project of the Center for Excellence in Regional Atmospheric Environment, CAS (No. CERAE201806).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.03.011.
| [1] |
S. Shwan, R. Nedyalkova, J. Jansson, et al., Ind. Eng. Chem. Res. 51 (2012) 12762-12772. DOI:10.1021/ie301516z |
| [2] |
R. Nedyalkova, S. Shwan, M. Skoglundh, L. Olsson, Appl. Catal. B:Environ. 138 (2013) 373-380. |
| [3] |
H. Chen, W.M.H. Sachtler, Catal. Today 42 (1998) 73-83. DOI:10.1016/S0920-5861(98)00078-9 |
| [4] |
P. Balle, B. Geiger, D. Klukowski, et al., Appl. Catal. B:Environ. 91 (2009) 587-595. DOI:10.1016/j.apcatb.2009.06.031 |
| [5] |
P. Balle, B. Geiger, S. Kureti, Appl. Catal. B:Environ. 85 (2009) 109-119. DOI:10.1016/j.apcatb.2008.07.001 |
| [6] |
S. Shwan, J. Jansson, L. Olsson, M. Skoglundh, Catal.Sci.Technol. 4 (2014) 2932-2937. DOI:10.1039/C4CY00236A |
| [7] |
M. Mauvezin, G. Delahay, B. Coq, et al., J. Phys. Chem. B 105 (2001) 928-935. DOI:10.1021/jp0021906 |
| [8] |
J. Perez-Ramirez, J.C. Groen, A. Bruckner, et al., J. Catal. 232 (2005) 318-334. DOI:10.1016/j.jcat.2005.03.018 |
| [9] |
J.A.Z. Pieterse, G.D. Pirngruber, J.A. van Bokhoven, S. Booneveld, Appl. Catal. B:Environ. 71 (2007) 16-22. DOI:10.1016/j.apcatb.2006.08.011 |
| [10] |
K. Jisa, J. Novakova, M. Schwarze, et al., J. Catal. 262 (2009) 27-34. DOI:10.1016/j.jcat.2008.11.025 |
| [11] |
S. Sklenak, P.C. Andrikopoulos, B. Boekfa, et al., J. Catal. 272 (2010) 262-274. DOI:10.1016/j.jcat.2010.04.008 |
| [12] |
S. Shwan, R. Nedyalkova, J. Jansson, et al., Top. Catal. 56 (2013) 80-88. DOI:10.1007/s11244-013-9933-4 |
| [13] |
H. Liu, J. Wang, T. Yu, S. Fan, M. Shen, Catal. Sci. Technol. 4 (2014) 1350-1356. DOI:10.1039/C3CY01026K |
| [14] |
L. Xu, C. Shi, B. Chen, et al., Microporous Mesoporous Mater. 236 (2016) 211-217. DOI:10.1016/j.micromeso.2016.08.042 |
| [15] |
D.S. Krivoruchenko, A.V. Kucherov, N.S. Telegina, et al., Russ. Chem. Bull. 63 (2014) 389-395. DOI:10.1007/s11172-014-0442-9 |
| [16] |
S. Jiang, R. Zhou, Fuel Process. Technol. 133 (2015) 220-226. DOI:10.1016/j.fuproc.2015.02.004 |
| [17] |
J. Li, L. Jia, W. Jin, F. Xia, J. Wang, Rare Metal Mater. Eng. 44 (2015) 1612-1616. DOI:10.1016/S1875-5372(15)30102-8 |
| [18] |
C. Pagis, A.R.M. Prates, D. Farrusseng, N. Bats, A. Tuel, Chem. Mater. 28 (2016) 5205-5223. DOI:10.1021/acs.chemmater.6b02172 |
| [19] |
E. Koohsaryan, M. Anbia, Chin. J. Catal. 37 (2016) 447-467. DOI:10.1016/S1872-2067(15)61038-5 |
| [20] |
L. Chen, X. Li, J.C. Rooke, et al., J. Mat. Chem. 22 (2012) 17381-17403. DOI:10.1039/c2jm31957h |
| [21] |
F. Xiao, L. Wang, C. Yin, et al., Angew. Chem. Int. Ed. 45 (2006) 3090-3093. |
| [22] |
K. Na, M. Choi, R. Ryoo, Microporous Mesoporous Mater. 166 (2013) 3-19. DOI:10.1016/j.micromeso.2012.03.054 |
| [23] |
P.N.R. Vennestrom, M. Grill, M. Kustova, et al., Catal. Today 168 (2011) 71-79. DOI:10.1016/j.cattod.2011.03.045 |
| [24] |
J. Ma, D. Weng, X. Wu, Z. Si, Z. Wu, Prog. Nat. Sci. 23 (2013) 493-500. DOI:10.1016/j.pnsc.2013.08.005 |
| [25] |
L. Ma, H. Chang, S. Yang, et al., Chem. Eng. J. 209 (2012) 652-660. DOI:10.1016/j.cej.2012.08.042 |
| [26] |
Y. Zhu, B. Chen, R. Zhao, et al., Catal. Sci. Technol. 6 (2016) 6581-6592. DOI:10.1039/C6CY00231E |
| [27] |
M. Jablonska, G. Delahay, K. Kruczala, et al., J. Phys. Chem. C 120 (2016) 16831-16842. DOI:10.1021/acs.jpcc.6b05692 |
| [28] |
H. Xia, H. Hu, S. Xu, K. Xiao, S. Zuo, Biomass Bioenergy 108 (2018) 426-432. DOI:10.1016/j.biombioe.2017.12.007 |
| [29] |
A.R.M. Prates, C. Pagis, F.C. Meunier, et al., Cryst.Growth Des. 18 (2018) 592-596. DOI:10.1021/acs.cgd.7b01635 |
| [30] |
A. Wang, Y. Wang, E.D. Walter, et al., J. Catal. 358 (2018) 199-210. DOI:10.1016/j.jcat.2017.12.011 |
| [31] |
S. Brandenberger, O. Krocher, A. Tissler, R. Althoff, Appl. Catal. B:Environ. 95 (2010) 348-357. DOI:10.1016/j.apcatb.2010.01.013 |
| [32] |
M.S. Kumar, M. Schwidder, W. Grunert, A. Bruckner, J.Catal. 227 (2004) 384-397. DOI:10.1016/j.jcat.2004.08.003 |
| [33] |
D.E. Doronkin, A.Y. Stakheev, A.V. Kucherov, et al., Top. Catal. 52 (2009) 1728-1733. DOI:10.1007/s11244-009-9327-9 |
| [34] |
M. Hoj, M.J. Beier, J.D. Grunwaldt, S. Dahl, Appl. Catal. B:Environ. 93 (2009) 166-176. DOI:10.1016/j.apcatb.2009.09.026 |
| [35] |
M.Y. Kim, K.W. Lee, J.H. Park, et al., Korean J. Chem. Eng. 27 (2010) 76-82. DOI:10.1007/s11814-010-0099-5 |
| [36] |
R.Q. Long, R.T. Yang, J. Catal. 194 (2000) 80-90. DOI:10.1006/jcat.2000.2935 |
| [37] |
Y. Xia, W. Zhan, Y. Guo, Y. Guo, G. Lu, Chin. J. Catal. 37 (2016) 2069-2078. DOI:10.1016/S1872-2067(16)62534-2 |
| [38] |
H. Pan, Y. Guo, H. Bi, Chem. Eng. J. 280 (2015) 66-73. DOI:10.1016/j.cej.2015.05.093 |
| [39] |
L. Ma, J. Li, H. Arandiyan, et al., Catal. Today 184 (2012) 145-152. DOI:10.1016/j.cattod.2011.10.007 |
| [40] |
L. Xu, C. Shi, Z. Zhang, et al., Microporous Mesoporous Mater. 200 (2014) 304-310. DOI:10.1016/j.micromeso.2014.04.034 |
| [41] |
P. Boron, L. Chmielarz, J. Gurgul, et al., Microporous Mesoporous Mater. 203 (2015) 73-85. DOI:10.1016/j.micromeso.2014.10.023 |
| [42] |
Q. Zeng, X. Bai, J. Zheng, J. Chen, R. Li, Chin. Chem. Lett. 22 (2011) 1103-1106. DOI:10.1016/j.cclet.2011.04.006 |
| [43] |
S. Yang, J. Li, C. Wang, et al., Appl. Catal. B:Environ. 117 (2012) 73-80. |
| [44] |
J.H. Baek, S.M. Lee, J.H. Park, et al., J. Ind. Eng. Chem. 48 (2017) 194-201. DOI:10.1016/j.jiec.2017.01.002 |
| [45] |
S. Shwan, E.C. Adams, J. Jansson, M. Skoglundh, Catal. Lett. 143 (2013) 43-48. DOI:10.1007/s10562-012-0940-z |
| [46] |
W. Su, H. Chang, Y. Peng, C. Zhang, J. Li, Environ. Sci. Technol. 49 (2015) 467-473. DOI:10.1021/es503430w |
| [47] |
E. Bourgeat-Lami, P. Massiani, F.D. Renzo, et al., Appl. Catal. A Gen. 72 (1991) 139-152. DOI:10.1016/0166-9834(91)85034-S |
| [48] |
S. Song, G. Wu, W. Dai, N. Guan, L. Li, Catal. Sci. Technol. 6 (2016) 8325-8335. DOI:10.1039/C6CY02124G |
| [49] |
S.A. Skarlis, D. Berthout, A. Nicolle, C. Dujardin, P. Granger, J. Phys. Chem. C 117 (2013) 7154-7169. DOI:10.1021/jp400717k |
| [50] |
Y. Peng, W. Yu, W. Su, X. Huang, J. Li, Catal. Today 242 (2015) 300-307. DOI:10.1016/j.cattod.2014.04.034 |
| [51] |
H. Huang, R.Q. Long, R.T. Yang, Appl. Catal. A:Gen. 235 (2002) 241-251. DOI:10.1016/S0926-860X(02)00268-5 |
| [52] |
P. Sazama, B. Wichterlova, S. Sklenak, et al., J. Catal. 318 (2014) 22-33. DOI:10.1016/j.jcat.2014.06.024 |
| [53] |
I. Melian-Cabrera, E.R.H. van Eck, S. Espinosa, et al., Appl. Catal. B:Environ. 203 (2017) 218-226. DOI:10.1016/j.apcatb.2016.10.019 |