 2019, Vol. 30
2019, Vol. 30 


b Hangzhou Institute of Innovative Medicine, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310018, China;
c Key Laboratory of Biomedical Engineering of the Ministry of Education, College of Biomedical Engineering & Instrument Science, Zhejiang University, Hangzhou 310027, China

Shuaifei Wang received his M.S. from the College of Pharmaceutical Science at Zhejiang University under the supervision of Prof. Daishun Ling. He currently focuses on the controlled synthesis of functional nanoparticles and their applications in tumor theranostics;

Hongwei Liao received his M.S. from the Zhejiang University School of Medicine under the supervision of Prof. Songmin Ying. He is currently a PhD candidate in College of Pharmaceutical Science at Zhejiang University under the supervision of Prof. Daishun Ling. He works on cell-nanoparticle interactions and biomedical applications of nanomaterials in cancer treatment;

Fangyuan Li received her M.S. (2011) from the Department of Biotechnology at The Catholic University of Korea. She obtained her Ph.D. (2015) from School of Chemical and Biological Engineering at Seoul National University. She joined the faculty of the College of Pharmaceutical Sciences at Zhejiang University in 2015 and currently working on the development of novel nano-platforms for medical diagnosis and therapy;

Daishun Ling received his Ph.D. (2013) from School of Chemical and Biological Engineering at Seoul National University. Later, he worked as a senior researcher at the Center for Nanoparticle Research, Institute for Basic Science. He joined the faculty of the College of Pharmaceutical Science at Zhejiang University in 2014. He currently focuses on the synthesis and assembly of functional nanoparticles for biomedical applications.
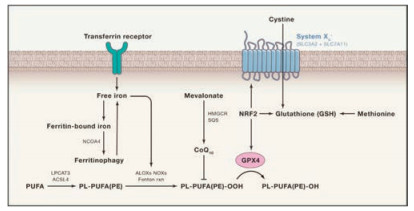
The nature of chemotherapies is to relieve the tumor burden of the patients by eliminating cancer cells via inducing cell death, mostly regulated cell death represented by apoptosis [1, 2]. Dozens of anticancer agents including clinically used ones kill cancer cells by promoting apoptosis directly or indirectly. However, as an endogenous barrier that must be subdued or bypassed before cellular transformation, apoptosis, in many cases, is compromised due to augmented antiapoptotic signaling or loss of proapoptotic mechanisms, rendering cancer cells resistant to many apoptosisinducing chemotherapies [3, 4]. Therefore, it is plausible totarget an alternative cell death pathway that obviates the need for an intact apoptosis signaling cascade. In recent years, a noncanonical form of regulated cell death, namely ferroptosis, has shown great promise to extend current apoptosis-based chemotherapies. Ferroptosis was first defined by Stockwell's group in 2012 as a cell death subroutine caused by an iron-dependent irreversible accumulation of lipid peroxides [5, 6]. Under normal conditions, the oxidized lipid arising from interactions with reactive oxygen species (ROS) generated by iron-dependent Fenton reaction can be converted to non-toxic lipid alcohols byglutathione peroxidase 4 (GPX4) (Fig. 1), whereas in the context of excess iron ions or dysfunctional GPX4, lipid peroxides can reach to a cytotoxic level that eventually initiates ferroptosis[7]. Since cancer cells are already heavily burdened with oxidative stress due to their highly active metabolic processes, which manifests as elevated hydrogen peroxide (H2O2) in the tumor microenvironment [8], it is anticipated that they will be hypersensitive to dysregulated uptake of iron ions causing ROS overproduction, and to GPX4 inactivation impairing cellular tolerance to oxidative damage[9, 10]. Hence, as a cell death pathway intimately involving oxidative stress, ferroptosis appears to be an attractive rationale for developing anticancer drugs [11]. Indeed, numerous studies have demonstrated the effectiveness of cancer-killing by inducing ferroptosis, which is mainly accomplished by elevating the intracellularROS levelsand inactivating the activity of GPX4 [12-14].

|
Download:
|
| Fig. 1. Pathways controlling ferroptosis. The ferroptosis intimately involves multiple pathways, including iron transportation, the amino acid metabolism, polyunsaturated fatty acid (PUFA) metabolism and the biosynthesis of glutathione. Free iron ions catalyze ROS production through Fenton reaction, leading to oxidation of phospholipid polyunsaturated fatty acids (PL-PUFA(PE)) into PL-PUFA (PE)-OOH. The PL-PUFA(PE)-OOH can be reduced to nontoxic alcohols by GPX4 under the normal circumstances. When there is excessive iron or GPX4 is inhibited, lipid peroxides will accumulate and eventually trigger ferroptosis. Copied with permission [7]. Copyright 2017, Elsevier. | |
{kind=link}
Interestingly, elevated intracellular ROS level is often observed as an attendant effect of some chemotherapeutic drugs such as doxorubicin and hydroxyurea [15, 16]. However, anticancer agents that act primarily by increasing ROS production are rarely seen, largely because of the lack of a tumor-specific target that can minimize damage to normal tissues [17]. With our increasing understanding of ferroptosis, a number of ROS-producing agents have now been tested for their ability to trigger cell death by ferroptosis, most of which are based on intracellular Fenton reaction that involves ferrous ion (Fe2+) or ferric iron (Fe3+)-mediated catalytic degradation of H2O2 into strong oxidative hydroxyl radical (HO·) [18-20]. Of note, to improve the tumor specificity of ferroptosis-inducing agents, tumor-responsive nanomaterials are frequently employed[21].Given the central role of iron in ferroptosis, much effort has been put into iron-based nanomaterials such as ferumoxytol [22], amorphous iron nanoparticles [23] and ironorganic frameworks [24], which effectively induce ferroptosis under experimental conditions.However, extremely low pH (2–4) and high Fe dose (75 mg Fe/kg) are required for optimal Fenton reaction to elicitferroptosis, rendering it very difficult for these agents to be used clinically as a standalone drug [18, 19, 23]. In addition, non-ferrous metals with multiple oxidation states, such as manganese [25] and copper [26], have also been used to design ferroptosis-inducing agents, demonstrating the flexibility of ferroptosis-based therapeutics. While the main strategy to exacerbate ROS burden is to apply iron-based nanomaterials, inactivation of GPX4 heavily relies on small molecules such as the erastin, sorafenib, and sulfasalazine. These agents can inhibit the cysteine/glutamate transporter system xc-, leadingto depletion of the GPX4 substrate glutathione (GSH) and thus GPX4 inhibition [27-29]. Besides, genetic manipulation of GPX4, ACSL4, p53, and SLC7A11 have also shown effective in mounting ferroptosis, though limited by a lack of delivery system that is both efficient and safe enough [30-33].
In this mini-review, we summarize the recent advances in ferroptosis-based anticancer treatments, with a focus on nanomaterials. We will put an emphasis on Fenton reaction that has been extensively exploited to generate massive intracellular ROS, and introduce the application of non-ferrous elements with multiple oxidation states to induce ferroptosis under a more physiologically relevant pH. Our work may extend the concept of iron-dependent ferroptosis to a more general metal-induced cell death, thus facilitating the future development of new strategies to overcome the limitations of current ferroptosis-based cancer therapies.
2. Fenton reaction-based nanomaterials for ferroptosisIn Fenton reaction, H2O2 is converted by Fe2+ or Fe3+ to highly oxidative ROS (Eqs. (1) and (2)), which is commonly used to catalyze the degradation of refractory organics [34]. Since the tumor microenvironment is characterized by low acidity and highlevel H2O2, it is expected that, by introducing a plethora of iron ions, Fenton reaction can be exploited in this context to generate intense oxidative stress [35]. Based on this, many iron-based agents, mostly nano-formulated, have been designed to release iron ions in response to the tumor microenvironment, which can promote overproduction of ROS by Fenton reaction. Consequently, the cancer cells accumulate a large amount of lipid peroxides and eventually undergo ferroptosis [18, 19]. Notably, some non-ferrous metal ions can also catalyze the generation of ROS from H2O2 in a Fenton-like reaction, and they exhibit some advantages over iron, as will be described in below [25, 26, 34].

|
(1) |

|
(2) |
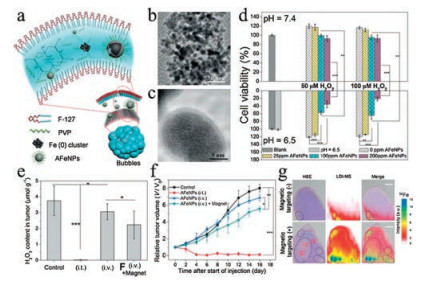
As shown in Eqs. (1) and (2), the concentration of iron ions is the determinant of the efficiency of Fenton reaction to generate ROS. Hence, it is reasonable to hijack this reaction by manipulating iron content using iron-based agents, which represents a practical strategy for increasing the oxidative stress of cancer cells to induce ferroptosis [36]. In fact, iron-based agents were already available before ferroptosis was characterized. Though not initially designed for cancer treatment, some of these early agents have manifested anticancer effects lately ascribed to the ability to induce ferroptosis [37]. A typical example is Ferumoxytol, which is an FDA-approved treatment for iron deficiency but later demonstrated to be able to cause tumor growth inhibition [22]. However, these agents lack tumor specificity and thus cannot be enriched in the tumor sites, limiting their therapeutic potency at a tolerable dose. In recent years, nanotechnologies that allow targeted delivery and tumor responsive drug release have offered a solution to this problem, prompting the design and synthesis of various iron-based nanomaterials for ferroptosis-based cancer therapy [12]. Zhang et al. synthesized amorphous Fe0 nanoparticles (AFeNPs) as a ferroptosis-inducing agent, which can rapidly release large amounts of Fe2+ because of the disordered random structure, greatly enhancing the Fenton reaction efficiency (Figs. 2a–c) [23]. As shown in Fig. 2d, the AFeNPs have higher cytotoxicity in the context of a lower pH and a higher H2O2 level, which are common features of the tumor microenvironment. Consistently, in vivo study also shows that administration of AFeNPs obviously causes a reduction in the H2O2 level in the tumor site, especially when administrated by the intratumoral injection (Fig. 2e). Most importantly, AFeNPs injected intravenously significantly suppress tumor growth, and even ablate tumors completely when given by intratumoral injection (Figs. 2f and g). In order to further improve the efficiency of intracellular Fenton reaction, Zhao et al. design a ferrouscysteine-phosphotungstate nanoagent to catalyze Fenton reaction under neutral pH, which displayed better therapeutic effects in vivo. Shen et al. designed a cisplatin-loaded Fe3O4/Gd2O3 nanoparticle and modified it with lactoferrin and RGD. Since the cisplatin can activate the activity of NADPH oxidases (NOXs) to generate more H2O2, the combination demonstrates higher efficiency than iron alone in generating excessive oxidative stress.

|
Download:
|
| Fig. 2. (a) Preparation of AFeNPs. (b, c) Low- and high-resolution TEM images of AFeNPs. (d) Growth inhibitory effects of the AFeNPs on MCF-7 cells under indicated pH and H2O2 concentration (n = 6, mean ± s.d., **P < 0.01, and ***P < 0.001). (e) Change of the H2O2 concentration in the tumor after different treatments (n = 3, mean ± s.d., *P < 0.05, and ***P < 0.001). (f) Tumor volume curve after different treatments (n = 5, mean ± s.d., *P < 0.05, **P < 0.01 and ***P < 0.001). (g) H&E staining images, LDI-MS 56Fe mapping, and merged images of tumor tissues after i.v. AFeNP injection with or without magnetic targeting. The circles highlight the isolated pockets. Copied with permission [23]. Copyright 2016, John Wiley & Sons. | |
{kind=link}
2.2. Manganese-based nanomaterials
As mentioned above, some non-ferrous metal ions can catalyze Fenton-like reaction to generate ROS, which therefore have been considered as an alternative besides iron to induce ferroptosis [38]. For example, Lin et al. designed manganese dioxide (MnO2)-coated mesoporous silica NPs (MS@MnO2 NPs) for cancer therapy [25]. Upon delivery to the cancer cells, the MnO2 can mediate the conversion of GSH, the natural intracellular antioxidants, to disulfide (GSSG), accompanied by the release of Mn2+. Subsequently, the Mn2+ can trigger the generation of HO· from H2O2 in the presence of HCO3-/CO2, increasing the intracellular oxidative stress to a tumoricidal extent (Fig. 3a). Through incubation with methylene blue (MB) that easily degrades upon HO· insult, it is manifested that MS@MnO2 NPs only catalyze the production of ROS in the presence of NaHCO3/CO2 (Fig. 3b), which improves tumor specificity since high-level NaHCO3/CO2 is common in the acidic tumor microenvironment. Meanwhile, the ROS-generating efficiency of MS@MnO2 NPs is also elevated under high GSH concentration (Fig. 3c), which is another feature of tumor cells. Moreover, the in vitro experiments have demonstrated a higher cytotoxicity of MS@MnO2 than MnCl2 (Fig. 3d). Most importantly, the in vivo study shows consistent potent anticancer effects of MS@MnO2 NPs (Figs. 3e and f), supporting the idea of using nonferrous metal in Fenton-like reaction to kill cancer cells by ferroptosis.

|
Download:
|
| Fig. 3. (a) The mechanism of MS@MnO2 NPs as a smart chemodynamic agent for MRI-monitored chemo-chemodynamic combination therapy. (b) UV/vis absorption spectra and photo (inset) of MB after degradation by the Mn2+-mediated Fentonlike reaction in different solutions. (c) MB degradation in the presence of H2O2 plus GSH-treated MS@MnO2 NPs. (d) Viability of U87MG cells after 24 h of incubation with MnCl2 (Mn2+), MS@MnO2 NPs, or MS@MnO2-CPT. (e) Tumor growth curves of mice after intravenous injection with different formulations. Each dose of Mn: 4.0 mg/kg for 2 × MS@MnO2 NPs group and 2.0 mg/kg for other groups. (f) TUNEL staining of tumor tissues from PBS and 2 × MS@MnO2 NPs groups. Scale bar: 200 mm. Copied with permission [25]. Copyright 2018, John Wiley & Sons. | |
{kind=link}
3. GPX4-inhibitory nanomaterials for ferroptosis
Recently, Stockwell's group has demonstrated that the GPX4 plays an essential role in the conversion of lipid peroxides to nontoxic lipid alcohols [39, 40]. Therefore, inactivating GPX4 is considered to be another approach to kill cancer cells via ferroptosis [41]. Based on this rationale, Zheng et al. designed a metal-organic network (MON) coated on the surface of polyethylenimine/p53 plasmid complex (MON-p53), which can kill the cancer cell by inducing ferroptosis (Fig. 4a) [24]. The preparation process of MON-p53 is shown in Fig. 4b. The polyethylenimine/p53 plasmid complex is prepared at first (Fig. 4c), which is then assembled as MON-p53 using the tannic-acid and ferric ions (Fig. 4d). When the MON-p53 is internalized into tumor cells, it will degrade and release iron ions that subsequently take part in the Fenton reaction, leading to elevated intracellular ROS levels (Fig. 4e). Meanwhile, the overexpression of the plasmid p53 can inhibit SLC7A11, a constitutive component of the cystine/glutamate antiporter system xc-, thus blocking the biosynthesis of GSH in cells. Without sufficient GSH supply, GPX4 can no longer protect the cells from the overwhelming ROS generated by MON-p53, which eventually leads to irreversible lipid peroxidation and cell death. Notably, the poisoned cell viability after treatments with MON-p53 can be rescued by ferroptosis inhibitors or lipid-soluble antioxidants, corroborating that MON-p53 induces cell death by ferroptosis (Fig. 4f). Additionally, in support of the ROS-generating capability of MON-p53, the intracellular levels of lipid peroxides, ROS and NADP+/NADPH, which indicate oxidative stress, are all evidently elevated after MON-p53 exposure (Fig. 4g). Moreover, the in vivo study has shown that the MON-p53 can significantly suppress tumor growth and prolong the survival time of tumor-bearing mice in comparison to PEI/p53 or MON alone (Figs. 4h and i).

|
Download:
|
| Fig. 4. (a) Schematic illustration for MON-p53 mediated anticancer therapy. (b) Schematic illustration for the preparation of MON-p53. TEM images of PEI/p53 plasmid complex (c) and (d) MON-p53. (e) Deduced the mechanism of MON-p53 induced ferroptosis. (f) Viability of HT1080 cells treated as indicated, the concentrations are as following: MON-p53 ± ferrostatin-1 (Fer, 100 nmol/L), Ac-DEVD-CHO (Apo, 50 μmol/L), Necrostatin-1 (Nec, 490 nmol/L), 3-methyladenine (Aut, 60 μmol/L), deferoxamine (DFO, 100 μmol/L), glutamic (Glu, 1 μmol/L), cystine (Cys, 1 μmol/L), glutathione (GSH, 1 μmol/L), sodium ascorbate (VC, 20μmol/L), vitamin E (VE, 20 μmol/L); and treated with PEI/p53 ± Fer (100 nmol/L), Apo (50 μmol/L). (g) LPO, ROS, and NADP+/NADPH content of HT1080 cells treated with ferric chloride (Fe(Ⅲ), 10μmol/L; Fe(Ⅱ), 25 μmol/L), PEI/p53, Fe(Ⅱ)-MON-p53, and Fe(Ⅲ)-MON-p53. (h) Survival curves of mice receiving injections of Era at a dose of 5 mg/kg, MONP at 5 mg/kg, and DNA at 0.375 mg/kg (n = 7) in HT1080 tumorbearing mice. (ⅰ) HT1080 tumor volume curves of mice during in the first 25 days. Copied with permission [24]. Copyright 2017, American Chemical Society. | |
{kind=link}
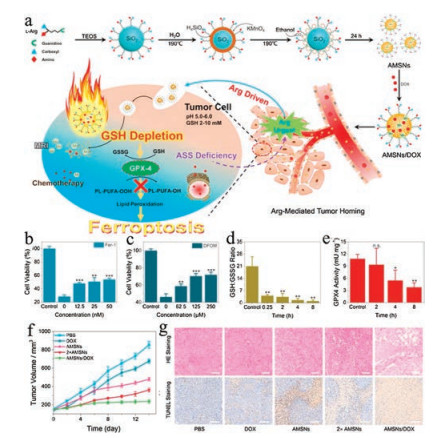
GSH is the predominant thiolated species in mammalian cells, which serves as a reducing agent lying at the core of the antioxidant systems of cells [42]. In the tumor microenvironment, the intracellular GSH concentration is much higher than extracellular levels [43, 44]. Previous studies have demonstrated that the GSH is an essential cofactor for GPX4 to eliminate the lipid peroxides in cells. Once the GSH pool is depleted, GPX4 will be inactivated, leading to ferroptosis [39, 45]. Our group has designed an arginine-rich manganese silicate nanobubbles (AMSNs) to promote ferroptosis by consuming the GSH pool and suppressing the activity of GPX4 (Fig. 5a) [46]. Consistent with the mechanism of action of AMSNs, the cytotoxicity induced by AMSNs can be relieved by the addition of ferroptosis inhibitor Fer-1and iron chelating agent desferrioxamine mesylate (DFOM) (Figs. 5b and c). The AMSNs effectively deplete GSH, as shown that the GSH level drops drastically within a short time after the treatment with AMSNs (Fig. 5d). Correspondingly, the activity of GPX4 is inhibited by AMSNs, though in a delayed manner (Fig. 5e). Importantly, this formulation exhibits potent in vivo efficacy in suppressing tumor growth and can be further strengthened when loaded with chemotherapeutic drugs (Figs. 5f and g).

|
Download:
|
| Fig. 5. (a) Schematic illustration of the designed synthesis of the arginine-rich manganese silicate nanobubbles (AMSNs) and the in vivo tumor homing after blood circulation. Viability of Huh7 cells treated with AMSNs plus Fer-1 (b) or DFOM (c). (d) GSH/GSSG level in Huh7 cells treated with AMSNs for different durations. (e) GPX4 activity measured at different times after treatment with AMSNs. (f) Tumorgrowth inhibitory effects of the different treatments. (g) H&E and TUNEL staining of tumor slices obtained from BALB/C mice after various treatments. (Scale bar: 100 μm). Copied with permission [46]. Copyright 2018, American Chemical Society. | |
{kind=link}
4. Other strategies for ferroptosis
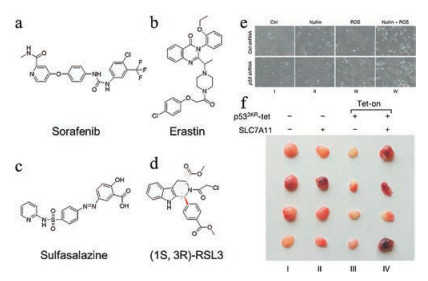
In addition to ROS-generating and GPX4 inactivating nanomaterials, small molecules and genetic manipulation targeting key factors in the ferroptosis pathway have also been tested for their anticancer effects [39, 47, 48]. Among a myriad of proteins involved in ferroptosis, system xc- may be the most studied target for small molecule-based intervention. System xc- is a cystine/glutamate antiporter transporting cysteine into cells, which is a rate-limiting step in the biosynthesis of GSH [49]. Small molecules including erastin, sorafenib and sulfasalazine can inhibit the function of system xc- and therefore impair GSH synthesis, leading to GPX4 inactivation and eventually ferroptosis (Figs. 6a–c) [12, 39, 50]. As to GPX4 itself, the small molecule (1S, 3R)-RSL3 can directly bind to it and inhibit its activity, which allows accumulation of lipid peroxides (Fig. 6d) [39, 47]. The well-known tumor suppressor p53 also plays a central role in ferroptosis. Genetic manipulation of p53 shows that p53 knockdown renders cancer cells resistant to oxidative stress, while its overexpression often synergizes with ferroptosis-inducing agents [51]. Jiang et al. observed that knockout of p53 protein could enhance the cell viability after the treatment of nutlin and ROS (Fig. 6e). SLC7A11 appears to be the downstream target of p53 to regulate ferroptosis, as shown that the p533KR-mediated tumor growth inhibition can be counteracted by the overexpression of SLC7A11 in a xenograft model (Fig. 6f) [30]. Recently, Doll et al. have identified ACSL4 as an integral component of the ferroptosis execution [52]. Notably, the expression of this protein is predictive of the sensitivity of basal-like breast cancer cells to ferroptosis. Since ACSL4 promotes ferroptosis in response to oxidative stress, overexpressing ACSL4 via plasmid DNA may confer a synergistic effect with other ferroptosis-inducing agents.

|
Download:
|
| Fig. 6. (a-d) Structures of the small-molecules used as ferroptosis-inducing agents in cancer therapy. (e) Bright field images of p53-proficient or -deficient U2OS cells treated with nutlin and ROS for 24 h. (f) SLC7A11 overexpression counteracts tumor growth suppression induced by p533KR in xenograft models. Copied with permission [30]. Copyright 2015, Springer Nature. | |
{kind=link}
5. Conclusions and perspectives
Since ferroptosis was first identified in 2012, it has been intensively studied for its mechanisms. A number of distinct features of ferroptosis have distinguished it from other cell death pathways, including but not limited to lipid peroxides accumulation, elevated Ptgs2, overexpression of Acyl-CoA synthetase long-chain family member 4 (ACSL4), and upregulated cation transport regulator-like protein 1 (CHAC1), these features are also used as biomarkers to examine ferroptosis. The evolving knowledge of how this new form of regulated cell death is executed lays the foundation for various strategies for the ferroptosis-based tumor therapy [5, 7, 39, 53, 54]. This mini-review summarized the development and application of various ferroptosis-inducing agents for cancer therapy in recent years, including the Fenton reaction-based nanomaterials, GPX4- based nanomaterials, small molecules, and gene technologies. The small molecules and gene technologies mainly act by impairing the GPX4-centered antioxidation mechanism of cancer cells, which consequently causes a failure to convert toxic lipid peroxides to nontoxic lipid alcohols. Though displaying potent anticancer effects, the small molecules inevitably have adverse effects on normal tissues because of the lack of specificity. In addition, short blood half-lives of small molecules in vivo also limit their therapeutic performance in clinical settings. As for gene therapy, safety concerns about offtarget effects and uncertain dose-effect relationship are still major obstacles to its further development [12].
Distinct from small molecules and gene technologies targeting the antioxidation mechanism, Fenton reaction-based nanomaterials exploit ferroptosis to kill cancer cells by generating excessive ROS. Up to now, iron-based nanomaterials are the most studied Fenton reaction-based ferroptosis-inducing agent, which can release iron ions to elevate the intracellular ROS burden. The plethora of intracellular ROS can overload the GPX4-mediated antioxidation activity, leading to irreversible accumulation of lipid peroxides and eventually cell death by ferroptosis. In order to improve their therapeutic efficiency, iron-based nanomaterials are usually used in combination with other ferroptosis-inducing agents, such as encapsulated H2O2 [55], lipid-hydroperoxide [56], and cisplatin [18].
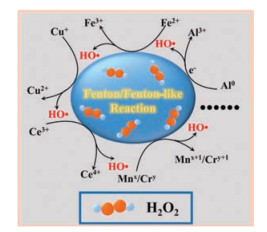
In principle, an ideal Fenton reaction element should have multiple oxidation states to form a redox cycle in the process of H2O2 decomposition to HO·. Although iron is very effective in catalyzing Fenton reaction, it requires a low pH value (pH 2-4) to reach the optimal catalytic efficiency, which is normally unavailable in the cellular context. Therefore, many non-ferrous elements with multiple oxidation states, such as the Mnx+/Mnx+1, Cu+/Cu2+, Al0/Al3+, Co2+/Co3+, Ce3+/Ce4+, Ruy+/Ruy+1, and Cr3+/Cr6+, have been investigated as highly efficient Fenton reaction reagents that can act under a biologically relevant pH (Fig. 7) [34]. Hence, the concept of ferroptosis could be extended as a type of cell death caused by an excess of various polyvalent metals beyond iron.

|
Download:
|
| Fig. 7. Schematic illustration for various metal ions with multiple oxidation states catalyzing the Fenton/Fenton-like reaction, in which H2O2 decomposes to highly oxidative reactive oxygen species. | |
{kind=link}
To conclude, exploiting Fenton reaction to kill cancer cells by inducing ferroptosis represents a brilliant strategy to fight cancer. Nanomaterials, primarily iron-based nanoagents, are the epitome of this strategy and exhibit potent therapeutic effects. Since nonferrous metals with multiple oxidation states are also capable of eliciting ferroptosis, we are now provided with even more options when devising anticancer tactics. With the advancement of nanotechnologies, more metal oxide nanoparticles or metal complexes will be designed and synthesized. These agents, either used alone or in combination with other ferroptosis-inducing therapeutics such as small molecules or siRNA, may greatly improve current cancer therapeutics.
AcknowledgmentsThis work was supported by the National Key Research and Development Program of China (No. 2016YFA0203600), the National Natural Science Foundation of China (Nos. 31822019, 91859116 and 51703195), the Fundamental Research Funds for the Central Universities (No. 2018QNA7020), the One Belt and One Road International Cooperation Project from Key Research and Development Program of Zhejiang Province (No. 2019C04024).
| [1] |
R. Rathore, J.E. McCallum, E. Varghese, A.M. Florea, D. Büsselberg, Apoptosis 22 (2017) 898-919. DOI:10.1007/s10495-017-1375-1 |
| [2] |
Y. Lu, Y. Wang, L. Miao, et al., Cancer Lett. 379 (2016) 32-38. DOI:10.1016/j.canlet.2016.05.025 |
| [3] |
A. Ashkenazi, W.J. Fairbrother, J.D. Leverson, A.J. Souers, Nat. Rev. Drug Discov. 16 (2017) 273-284. DOI:10.1038/nrd.2016.253 |
| [4] |
R.W. Johnstone, A.A. Ruefli, S.W. Lowe, Cell 108 (2002) 153-164. DOI:10.1016/S0092-8674(02)00625-6 |
| [5] |
S. Dixon, K. Lemberg, M. Lamprecht, et al., Cell 149 (2012) 1060-1072. DOI:10.1016/j.cell.2012.03.042 |
| [6] |
S. Rachid, S.J. Dixon, W. Jianlin, et al., J. Am. Chem. Soc. 136 (2014) 4551-4556. DOI:10.1021/ja411006a |
| [7] |
B.R. Stockwell, J.P. Friedmann Angeli, H. Bayir, et al., Cell 171 (2017) 273-285. DOI:10.1016/j.cell.2017.09.021 |
| [8] |
Q. Chen, L. Feng, J. Liu, et al., Adv. Mater. 28 (2016) 7129-7136. DOI:10.1002/adma.201601902 |
| [9] |
V.E. Kagan, G. Mao, F. Qu, et al., Nat. Chem. Biol. 13 (2016) 81-90. |
| [10] |
S.J. Dixon, B.R. Stockwell, Nat. Chem. Biol. 10 (2014) 9-17. DOI:10.1038/nchembio.1416 |
| [11] |
B.J. Crielaard, T. Lammers, S. Rivella, Nat. Rev. Drug Discov. 16 (2017) 400-423. DOI:10.1038/nrd.2016.248 |
| [12] |
Z. Shen, J. Song, B.C. Yung, Adv. Mater. 30 (2018) 1704007. DOI:10.1002/adma.v30.12 |
| [13] |
K. Shimada, R. Skouta, A. Kaplan, et al., Nat. Chem. Biol. 12 (2016) 497-503. DOI:10.1038/nchembio.2079 |
| [14] |
S.E. Kim, L. Zhang, K. Ma, et al., Nat. Nanotechnol. 11 (2016) 977-985. DOI:10.1038/nnano.2016.164 |
| [15] |
S.Y. Kim, S.J. Kim, B.J. Kim, et al., Exp. Mol. Med. 38 (2006) 535-545. DOI:10.1038/emm.2006.63 |
| [16] |
B.W. Davies, M.A. Kohanski, L.A. Simmons, et al., Mol. Cell 36 (2009) 845-860. DOI:10.1016/j.molcel.2009.11.024 |
| [17] |
S. Zhang, X. Liu, T. Bawa-Khalfe, et al., Nat. Med. 18 (2012) 1639-1642. DOI:10.1038/nm.2919 |
| [18] |
Z. Shen, T. Liu, Y. Li, et al., ACS Nano 12 (2018) 11355-11365. DOI:10.1021/acsnano.8b06201 |
| [19] |
P. Zhao, Z. Tang, X. Chen, et al., Mater. Horiz. 6 (2019) 369-374. DOI:10.1039/C8MH01176A |
| [20] |
Z. Tang, H. Zhang, Y. Liu, et al., Adv. Mater. 29 (2017) 1701683. DOI:10.1002/adma.201701683 |
| [21] |
L. Yue, J. Wang, Z. Dai, et al., Bioconjugate Chem. 28 (2017) 400-409. DOI:10.1021/acs.bioconjchem.6b00562 |
| [22] |
S. Zanganeh, G. Hutter, R. Spitler, et al., Nat. Nanotechnol. 11 (2016) 986-994. DOI:10.1038/nnano.2016.168 |
| [23] |
C. Zhang, W. Bu, D. Ni, et al., Angew. Chem. Int. Ed. 128 (2016) 2141-2146. DOI:10.1002/ange.201510031 |
| [24] |
D.W. Zheng, Q. Lei, J.Y. Zhu, et al., Nano Lett. 17 (2017) 284-291. DOI:10.1021/acs.nanolett.6b04060 |
| [25] |
L.S. Lin, J. Song, L. Song, et al., Angew. Chem. Int. Ed. 130 (2018) 4996-5000. DOI:10.1002/ange.201712027 |
| [26] |
B. Ma, S. Wang, F. Liu, et al., J. Am. Chem. Soc. 141 (2019) 849-857. DOI:10.1021/jacs.8b08714 |
| [27] |
C. Louandre, I. Marcq, H. Bouhlal, et al., Cancer Lett. 356 (2015) 971-977. DOI:10.1016/j.canlet.2014.11.014 |
| [28] |
W.S. Yang, K.J. Kim, M.M. Gaschler, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 4966-4975. DOI:10.1073/pnas.1603244113 |
| [29] |
J.C. Reed, M. Pellecchia, Cell 149 (2012) 963-965. DOI:10.1016/j.cell.2012.05.009 |
| [30] |
L. Jiang, K. Ning, T. Li, et al., Nature 520 (2015) 57-62. DOI:10.1038/nature14344 |
| [31] |
I. Ingold, C. Berndt, S. Schmitt, et al., Cell 69 (2017) 423-434. |
| [32] |
K. D'Herde, D.V. Krysko, Nat. Chem. Biol. 13 (2017) 4-5. DOI:10.1038/nchembio.2261 |
| [33] |
Y. Xie, W. Hou, X. Song, et al., Cell Death Differ. 23 (2016) 369-379. DOI:10.1038/cdd.2015.158 |
| [34] |
A.D. Bokare, W. Choi, J. Hazard. Mater. 275 (2014) 121-135. DOI:10.1016/j.jhazmat.2014.04.054 |
| [35] |
Z. Tang, Y. Liu, M. He, W. Bu, Angew. Chem. Int. Ed. 58 (2019) 946-956. DOI:10.1002/anie.201805664 |
| [36] |
A. Tarangelo, S.J. Dixon, Nat. Nanotechnol. 11 (2016) 921-922. DOI:10.1038/nnano.2016.199 |
| [37] |
P.A. Ma, H. Xiao, C. Yu, et al., Nano Lett. 17 (2017) 928-937. DOI:10.1021/acs.nanolett.6b04269 |
| [38] |
T. Valdés-Solís, P. Valle-Vigón, S. Álvarez, G. Marbán, A.B. Fuertes, Catal. Commun. 8 (2007) 2037-2042. DOI:10.1016/j.catcom.2007.03.030 |
| [39] |
W.S. Yang, R. Sriramaratnam, M. Welsch, et al., Cell 156 (2014) 317-331. DOI:10.1016/j.cell.2013.12.010 |
| [40] |
J.P. Friedmann Angeli, M. Schneider, B. Proneth, et al., Nat. Cell Biol. 76 (2014) 1180-1191. |
| [41] |
M.J. Hangauer, V.S. Viswanathan, M.J. Ryan, et al., Nature 551 (2017) 247-250. DOI:10.1038/nature24297 |
| [42] |
J.M. Estrela, A. Ortega, E. Obrador, Crit. Rev. Clin. Lab. Sci. 43 (2006) 143-181. DOI:10.1080/10408360500523878 |
| [43] |
X. Ling, X. Chen, I.A. Riddell, et al., Nano Lett. 18 (2018) 4618-4625. DOI:10.1021/acs.nanolett.8b01924 |
| [44] |
J. Wang, X. Sun, W. Mao, et al., Adv. Mater. 25 (2013) 3670-3676. DOI:10.1002/adma.v25.27 |
| [45] |
S. Ma, E.S. Henson, Y. Chen, S.B. Gibson, Cell Death Dis. 7 (2016) e2307. DOI:10.1038/cddis.2016.208 |
| [46] |
S. Wang, F. Li, R. Qiao, et al., ACS Nano 12 (2018) 12380-12392. DOI:10.1021/acsnano.8b06399 |
| [47] |
T. Krainz, M.M. Gaschler, C. Lim, et al., ACS Cent. Sci. 2 (2016) 653-659. DOI:10.1021/acscentsci.6b00199 |
| [48] |
X. Sun, Z. Ou, R. Chen, et al., Hepatology 63 (2016) 173-184. DOI:10.1002/hep.28251 |
| [49] |
P.N. Mccormick, H.E. Greenwood, M. Glaser, et al., Cancer Res. 79 (2019) 853-863. DOI:10.1158/0008-5472.CAN-18-2634 |
| [50] |
W.S. Yang, B.R. Stockwell, Trends Cell Biol. 26 (2016) 165-176. DOI:10.1016/j.tcb.2015.10.014 |
| [51] |
S.J. Wang, D. Li, Y. Ou, et al., Cell Rep. 17 (2016) 366-373. DOI:10.1016/j.celrep.2016.09.022 |
| [52] |
S. Doll, B. Proneth, Y.Y. Tyurina, et al., Nat. Chem. Biol. 13 (2016) 91-98. |
| [53] |
H. Yuan, X. Li, X. Zhang, R. Kang, D. Tang, Biochem. Biophys. Res. Commun. 478 (2016) 1338-1443. DOI:10.1016/j.bbrc.2016.08.124 |
| [54] |
Y. Xie, S. Zhu, X. Song, et al., eLife 3 (2014) e02523. DOI:10.7554/eLife.02523 |
| [55] |
W.P. Li, C.H. Su, Y.C. Chang, Y.J. Lin, C.S. Yeh, ACS Nano 10 (2016) 2017-2027. DOI:10.1021/acsnano.5b06175 |
| [56] |
Z. Zhou, J. Song, R. Tian, et al., Angew. Chem. Int. Ed. 129 (2017) 6492-6496. |