 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Bioreactor Engineering, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, China

Professor Youjun Yang got his bachelor's degree from the University of Science and Technology of China (USTC) and PhD from Louisiana State University (LSU) under the guidance of Prof. Robert M. Strongin, with a focus on xanthene and benzoxanthene synthesis. He then did postdoctoral study with Prof. Eric V. Anslyn in the University of Texas at Austin. In 2010, he started his independent career in the School of Pharmacy of East China University of Science and Technology (ECUST) in Shanghai. He was appointed as a full professor in 2015. The theme of his research is the development of dyes and probes for bioimaging and theranostics. His group has developed high-performance dyes (EC dyes) with absorption and emission wavelengths in the deep NIR spectral region (800 nm and beyond), novel Hill-type pH fluorescent probes (with a sharp acid-base transition width of ~0.5-1.2 pH unit), xanthene based potent antibiotic compounds (effective toward many high-priority drug-resistance pathogens), and novel molecular tools to study the implications of various substrates (NO/NO+, Hg2+/MeHg+, OONO-/ HOONO, Sarin, radicals, etc.) in human health and diseases. The awards he has received include the Excellent Young Scholar of the National Science Foundation of China (NSFC), the Czarnik Emerging Investigator award of the Molecular Sensors and Molecular Logic Gate symposium (MSMLG), the Rising-Star award of Shanghai Municipal of Science and Technology Commission (SMSCT), and the Presidential award of East China University of Science and Technology.
Infrared refers to the light beyond the red edge of our visual perception. "Near-infrared" or NIR is a routinely encountered term in the literature, to refer the blue end of the infrared spectral region [1-7]. Our eyes do not sensitively respond to the light beyond 700 nm. So, 700 nm is set as the blue edge of the NIR by the CIE [8]. Nevertheless, 780 nm is regarded as the blue edge of NIR by some others, e.g., the ISO [9]. Some others may deem 750 nm [10] or 800 nm [11] as the blue edge. The designation of the red edge of the NIR is as diverse, e.g., 900 nm or 950 nm in bioimaging, 1000 nm in material sciences, 1400 nm by the CIE, 1700 nm in bioimaging, 3000 nm by the ISO, 5000 nm in astronomy [12-16].
In the past few decades, fluorescence-based imaging techniques has gradually become indispensable for basic research [1, 17-21], translational studies and clinical theranostics [22-29]. The predominant biological pigments, e.g., hemoglobin and oxygenated hemoglobin, do not appreciably absorb the light beyond 650 nm. Light around 950 nm can be absorbed by water or lipids [30]. Therefore, the spectral range of 650–950 nm is transparent to the biological system, ideal for tissue or in vivo imaging and has been designated as the "biological window", "optical window" or "theranostic window" [15, 31, 32]. Since 2009, researchers [33, 34] pioneered and started to promote the use of a longer spectral region, i.e., 1000–1700 nm, for in vivo small animal imaging, and named this region "NIR-Ⅱ" [35-38], which has received wide acceptance by the community. The NIR-Ⅱ is further dissected into three sub-regions. The range of 1350–1500 nm is of limited utility for imaging because water vibronically absorbs there. Currently, the use of the bluer end (NIR-Ⅱa), i.e., 1000–1350 nm, is more popular due to the relative availability of materials emitting in this region. The use of the redder region (NIR-Ⅱb), i.e., 1500–1700 is less exploited. At the same time, "NIR-Ⅱ" and "NIR-Ⅲ" were occasionally used to refer to 1000–1350 nm and 1550–1870 nm respectively [39]. The NIR in this manuscript would refer to the spectral region of 650–1700 nm.
"NIR" materials are the materials spectrally active in the NIR region and could be of organic [28], inorganic [40], polymeric [41], and biological [42] in nature. The primary focus of this tutorial is organic NIR materials and hence "dyes" are generally used throughout the manuscript. NIR-absorbing dyes refer to dyes with an absorption maximum in the NIR region. NIR-emitting dyes are those with an emission maximum in the NIR region, and may be used interchangeably with the term "NIR fluorophores". NIR-emitting dyes (or NIR fluorophores) do not necessarily exhibit an absorption maximum in the NIR region. They could also be ones absorbing in the visible spectral region, but emitting in the NIR spectral region. Analogously, NIR-absorbing and NIR-emitting dyes could be used to emphasize that both the absorption and emission maxima fall in the NIR spectral region.
NIR dyes are not recent inventions. The first NIR-absorbing dye was first prepared by Piccard et al. in 1913 [43]. Polymethine cyanine dyes absorbing up to 1300 nm were made available in the early 1930s [44]. Originally, they were used in photography. Nowadays, their applications are widespread and encompass lasing, information storage, display, xeroxing and printing, occupational protection, solar cells, disease diagnosis, and medical treatments [45].
Fluorescence based techniques have become routine tools for chemistry, biology and medicine [46]. NIR light penetrate deeper in tissue as a result of reduced absorption and scattering. Recent years have witnessed a paradigm shift to the use of NIR fluorescent dyes for tissue or in vivo imaging. Despite the colossal need, NIR dyes are rare and the volume of related literature is limited, compared to the ones active in the visible spectrum. This is because their development, especially the high-performance ones, is challenging. First, to absorb NIR light, NIR dyes exhibit a smaller HOMO-LUMO gap than the visible dyes. In principle, this can be accomplished by raising the HOMO or lowering the LUMO of a visible dye. Yet, this is by no means easy. Second, the chemostabilities of NIR dyes are intrinsically poor, as a result of a small HOMO-LUMO gap. A low-lying LUMO orbital leads to susceptibility to nucleophilic attack and reduction, while a high-lying HOMO orbital leads to susceptibility to oxidation. On top of that, photostability of most NIR dyes are notoriously poor, as well. They may readily isomerize or decompose upon photoexcitation. NIR dyes without suitable stability have a slim practical significance. Third, fluorescence quantum yield of a NIR dyes is typically limited, owning to fast internal conversion kinetics, as a result of small HOMO-LUMO bandgap and poor structural rigidity.
Indocyanine Green (ICG) is without doubt the most widely accepted NIR dye for biomedical use [47]. It is a polymethine cyanine dye first prepared in 1930's for NIR photography. In 1957, it was tested for medical diagnostic applications and soon approved by FDA [48]. Nowadays, the medical use of ICG is widespread to include lymphangiography, intra-operative lymph node identification, tumor imaging, superficial vascular imaging, and marking ischemic tissues [49]. A literature survey with the key word "Indocyanine Green" yielded 8824 hits in the Web of Science.
ICG is an overall good NIR dye [50]. Its maximal absorption and emission wavelengths fall well within the NIR spectral region, at 780 nm and 813 nm in water respectively. It is a very strong absorber with a molar extinction coefficient of 113, 790 L mol-1 cm-1. It is also a NIR emitter with a notable fluorescence quantum yield of 13% in MeOH. However, it is not without limitation. ICG tends to aggregate in water at high concentration with a blueshifted absorption maximum to around 700 nm [51]. Over time, ICG aggregates with a new absorption maximum of 900 nm could appear [52]. Aggregation tremendously influences its optical properties and severely hampers its applications where quantitative measurements are needed. Second, it is not stable. Binding to plasma proteins, especially lipoproteins, redshifts the absorption peak of ICG to 805 nm, and yielding enhanced fluorescence intensity [53]. The decomposition of diluted aqueous solutions of ICG has been well documented by the apparent gradual decrease of its optical density, and exposure to light accelerates the photodegradation of ICG in a self-sensitized photo oxidation fashion, yielding several carbonyl decomposition compounds [54]. The conjugative backbone of ICG is prone to attack by nucleophilic reagents, including reactive oxygen/ nitrogen species (ONOO-, singlet oxygen) [55-59], intracellular thiols (cysteine, glutathione) and cyanide etc. [60-64]. Therefore, there is an ongoing and relentless wave of efforts toward development of bright, stable, biocompatible and longer-wavelength NIR fluorescent dyes.
2. Design rationale of NIR absorbing and emitting dyesThe design of a novel high-performance fluorescent NIR dye is a four-step endeavor: 1) choosing an electronic push-pull (D-π-A) scaffold, 2) band-gap manipulation by judicious substitution, 3) structural rigidification, and 4) steric protection.
2.1. The necessity of a D-π-A scaffoldThe relationship between the color and the constitution has been the central question of the field since the beginning of the synthetic dye era. Understandings toward this topic have been summarized in various seminal monographs [65-67]. Audience of this review is also recommended to go over the aforementioned sources for an in-depth discussion of the topic.
In short, light absorption by an organic molecule promotes the electronic transition from two different molecular orbitals. If such an organic molecule is a saturated compound, e.g., hydrocarbons, such a photo-promoted electronic transition has to be σ-σ*, which will result in breakage of the corresponding σ bond and formation of two radical species. In other words, this molecule is not photostable and cannot be possibly used as a dye. This is the reason why all dyes bear an unsaturated backbone.
The smallest unsaturated organic molecule is ethylene. It has a HOMO-LUMO band gap of 6.7 eV and absorbs deep UV light at 185 nm [68]. For a molecule to render color, it has to be able to absorb visible light (380–780 nm). Conjugation lowers the LUMO and raises HOMO simultaneously and therefore is effective to achieve longer wavelength absorption. For example, β-carotene is a polyene containing 11 conjugated double bonds and absorbs maximally at 450 nm [69]. Further extension of the length of the conjugated system can indeed further render a bathochromic shift, however only to a less appreciated extend, as a result of "effective conjugated length" [70]. It has been showcased that a 50-ene has a maximal absorption wavelength of 603 nm, and a 100-ene at 613 nm. Polyacetylene does not absorb beyond 650 nm [71]. Clearly, polyene is not the way to follow in search for a dye absorbing and emitting beyond the red-edge of the visible range.
Instead of extending the degree of conjugation in a one-dimensional fashion, can 2-D conjugation extension cost-effectively promote the absorption wavelength to 650 nm and beyond? Many of the molecules in Fig. 1 have not yet become available. Nevertheless, the trend is obvious that NIR absorption can be achieved when the p-conjugated system grows fairly large [72].However, such a 2D-conjugated sheet-like structure is prone to stacking, especially in aqueous medium.

|
Download:
|
| Fig. 1. Polyenes and polyaromatic hydrocarbons. | |
At the very early days of the synthetic dye chemistry, it has been noted that the presence of electron-donating group (i.e., donor, or push) and electron-withdrawing group (i.e., acceptor or pull) could induce a bathochromic shift of the absorption [72].
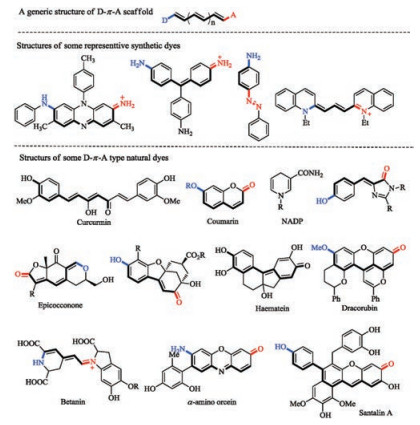
This is intuitive from a perspective of molecular orbital theory. The donor group can raise the HOMO while the acceptor group lowers the LUMO, leading to the decrease of the HOMO-LUMO band gap. The effect is reinforced when the donor and the acceptor are substituted to the conjugated system in such a fashion that electron delocalization from the donor to the acceptor, i.e., Internal Charge Transfer (ICT), is feasible. Such a molecular system is often referred to as a donor-π-acceptor (D-π-A), or an electronic pushpull system. Though exceptions exist especially with some very small structures, D-π-A has been firmly established as a viable guideline for development of novel dye scaffolds. Mauve, the dye that started the fine chemical industry, is such a D-π-A type chromophore. So are the azo dyes, triarylmethane dyes, cyanine dyes and many others from the early times of the synthetic dye era. Interestingly, many natural dyes also employ a D-π-A scaffold to impart color (Fig. 2) [73-75].

|
Download:
|
| Fig. 2. Structures of the generic D-π-A scaffold, some early synthetic coal-tar dyes and structures of the natural dyes. | |
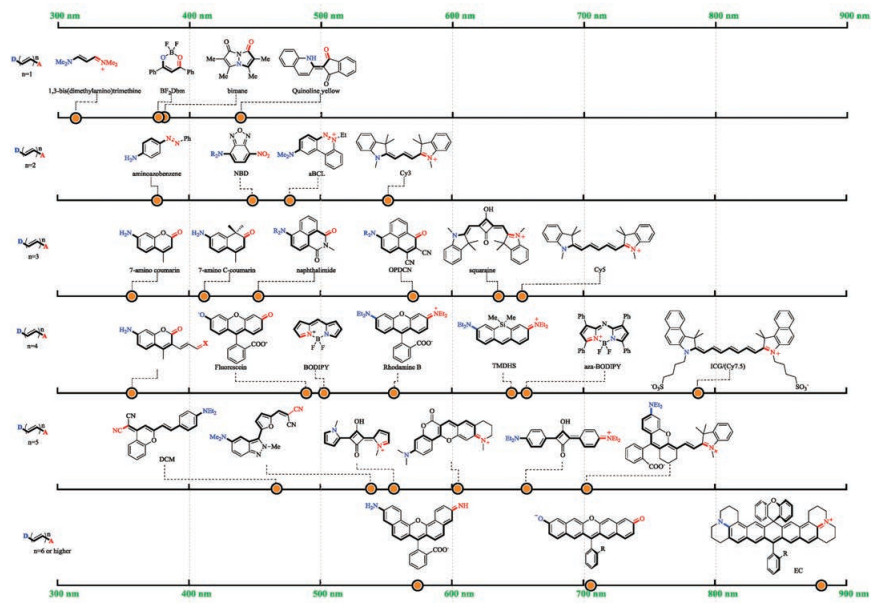
Though a longer D-π-A scaffold does not necessarily lead to a redder absorption wavelength, an NIR dye warrants the use of a large scaffold. As the potential choices of donor, π-conjugative backbone and acceptor become abundant, numerous more D-π-A type dyes have been created. Some classic scaffolds with a D-π-A principle are summarized (Fig. 3) [76-91]. The D-π-A scaffold of the same length does not necessarily lead to the same absorption wavelength. For example, both aminoazobenzene and Cy3 have a 7-atom D-π-A scaffold. Yet, the former absorbs at 360 nm and the latter at 560 nm. The maximal absorption wavelengths of various 9-atom D-π-A scaffolds fall in a rather broad range of ca. 350– 650 nm. The absorption wavelengths of 11-atom or 13-atom scaffolds are scattered in an even broader range.

|
Download:
|
| Fig. 3. Notable D-π-A type fluorescent dyes. | |
Two intuitive trends are obvious for D-π-A type dyes (Table 1). First, the stronger the electronic delocalization between the donor and the acceptor, the longer the absorption wavelength bathochromically shift. Since nitrogen is more electron-donating than oxygen due to a smaller electronegativity, a long wavelength dye typically employ an amino group as its electron donor. Analogously, iminium is more popular than carbonyl because it is a more potent electron acceptor. Merocyanine is not an ideal D-π-A scaffold if long-wavelength absorption is in pursuit. Second, with the donor and the acceptor groups remain unchanged, a longer conjugative backbone leads to a longer wavelength absorption. Both of these trends have been experimentally confirmed [66].
|
|
Table 1 Structures and absorption maxima (nm) of various simple D-π-A type scaffolds. |

{kind=link}
{kind=link}
{kind=link}
2.2. Further modulation of the HOMO-LUMO gap of a D-π-A scaffold
The absorption wavelength of a D-π-A scaffold could be further modulated by judicious installation of substituents of either electron-donating or electron-withdrawing nature. The effect could be readily predicted with the help of some sophomore organic chemistry.
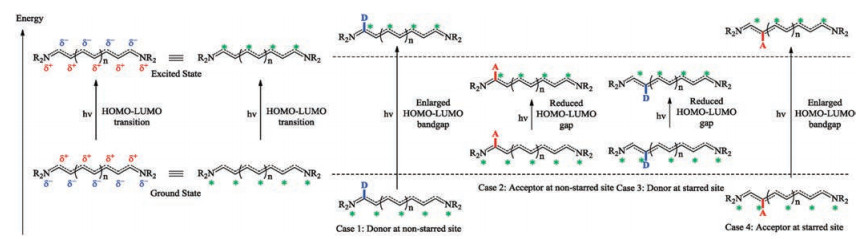
The Lewis structure is the way to keep track of the connections of various atoms within a given compound by every practitioner of organic chemistry. However, it is not without limitation, especially when electronic delocalization exists in the molecule. The resonance theory was introduced as a remedy. For example, the three major-contributing resonant structures are drawn for acetate (Fig. 4a). The implication of these three structures is that the negative charge does not localize to any specific one of the two oxygen atoms, but is delocalized between the two oxygen atoms. Therefore, each oxygen atom carries a partial negative charge (δ-). Similarly, it can be inferred that a partial positive (δ+) dwells at the carbonyl carbon atom. With the resonance theory, it can be concluded that the carboxylate, a D-π-A scaffold, exhibiting a unique alternating δ+/δ- feature. In the seminal book by Griffiths [66], these atoms with partial negative charges are labeled by a sign of star (*). Analogously, a D-π-A type cyanine dye can be drawn in different forms shown in Fig. 4b.

|
Download:
|
| Fig. 4. Alternative ways to represent the electronic delocalization in a D-π-A type scaffold. | |
{kind=link}
Absorption of a photon is accompanied by an electronic transition from a low-lying orbital to a high-lying empty orbital. Concomitantly, the molecule is promoted from the ground state to a higher energy state, i.e., the excited state. The D-π-A scaffold in its excited state has a different electronic distribution. For example, HOMO-LUMO transition induces the reversal of the electronic distribution. The atoms carrying partial negative charges in the ground state (or the starred atoms) now carries a partial positive charge and vice versa. With this knowledge, the impact of a substituent to the absorption wavelength of a D-π-A type dye is readily comprehensible (Fig. 5). The non-starred sites are electron deficient sites, which are stabilized by the presence of electron donating groups and destabilized by the electron withdrawing groups. Analogously, the starred sites are electron rich sites, which are stabilized by electron withdrawing groups. Analysis here is qualitative and exceptions could very well exist. However, when used as a general framework, the following set of rules could be of help for design of NIR dyes:

|
Download:
|
| Fig. 5. The impact of substitution on the HOMO-LUMO band-gap of the D-π-A scaffold. | |
{kind=link}
(a) An electron donating group at non-starred site results in stabilization of the ground state and the destabilization of the excited state. Hence the HOMO-LUMO energy gap increases and the absorption wavelength shifts to the shorter wavelength spectral region.
(b) An electron withdrawing group at non-starred site results in destabilization of the ground state and the stabilization of the excited state. Hence the HOMO-LUMO energy gap decreases and the absorption wavelength shifts to the longer wavelength spectral region.
(c) An electron donating group at starred site results in destabilization of the ground state and the stabilization of the excited state. Hence the HOMO-LUMO energy gap decreases and the absorption wavelength shifts to the longer wavelength spectral region.
(d) An electron withdrawing group at starred site results in stabilization of the ground state and the destabilization of the excited state. Hence the HOMO-LUMO energy gap increases and the absorption wavelength shifts to the shorter wavelength spectral region.
2.3. Rigidification of a D-π-A scaffoldExtension of the length of the conjugative backbone promotes the bathochromic shift of the absorption wavelength of a dye. However, as the backbone grows longer, it becomes increasingly flexible. The structural flexibility is particularly detrimental to the fluorescence quantum yield, enabling rotational or vibrational deactivation of the excited state of a dye. Rigidification of the D-π-A scaffold is therefore an indispensable step to the development of bright and stable NIR fluorescent dyes. Rigidification also promotes the stability of a dye.
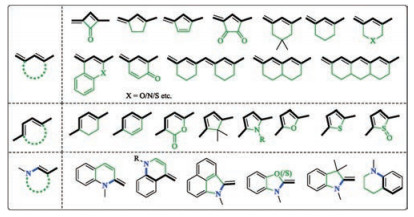
Though the D-π-A scaffolds in this review have typically been drawn in a way that the two adjacent double bonds are trans to each other. They can actually adopt either trans- or cis- configuration. A trans diene can be rigidified when locked in a ring. Such a ring can be four-membered, five-membered or six-membered. Examples are summarized in Fig. 6 [92-108]. When one ring does not provide sufficient degree of rigidification, two or more rings could be incorporated onto the conjugative backbone. These rings can either be separated or annulated. The cis-diene or the electron-donating enamine moiety can be similarly rigidified by locking into a ring. The ring is typically five-membered or six-membered.

|
Download:
|
| Fig. 6. Rigidification methods of the D-π-A scaffold. | |
{kind=link}
If an aromatic ring is resulted by ring-formation, the four electrons of the diene are now involved in aromatic ring current and become more localized (Fig. 7a). Electronic delocalization through this aromatic ring is less efficient. As a consequence, the two bonds connecting this aromatic ring to the remaining part of the D-π-A system have more single-bond characters and more readily rotate. So, the whole system is actually less rigid and such a system is usually not very fluorescent. To address this difficulty, the entire D-π-A backbone can be polycyclically rigidified, as exemplified with the structures of coumarin and fluorescein (Fig. 7b).

|
Download:
|
| Fig. 7. Rigidification of the cis-diene via aromatic rings. | |
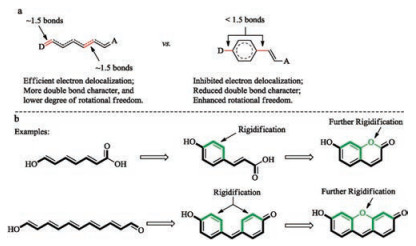{kind=link}
2.4. Steric protection of the D-π-A scaffold
An NIR dye has a low-lying LUMO orbital and therefore are intrinsically prone to nucleophilic attack. Unfortunately, the biological milieu has a plethora of nucleophilic species, e.g., H2O, sulfide, superoxide, hypochlorite and peroxynitrite, etc. While nucleophilic attack at the conjugative backbone of a D-π-A scaffold by hydroxide and sulfide is reversible and does not lead to permanent destruction of the dye, nucleophilic attack by superoxide, hypochlorite or peroynitrite does result in oxidative cleavage of the conjugative backbone and should be mitigated [55, 58, 109]. Also, a long and flexible conjugative backbone is prone to oxidation by singlet oxygen [110]. A viable approach is to protect the most electrophilic sites of a NIR dye by installation of sterics, which blocks the trajectories of incoming nucleophiles and protects the NIR dye in a kinetic fashion. Also, sterically protected NIR dyes cannot readily aggregate via π-π stacking.
The nature of sterics can be supramolecular or molecular. The installation of a supramolecular steric can be quite facile. The D-π-A backbone can be threaded into a macrocycle [111-115], embedded in a polymetric matrix [116, 117], or encased in an inert cavity [118-120]. This is a fascinating approach, yet not without limitation. First, the final supramolecular dye/sterics complex is a much larger scaffold than the original D-π-A scaffold. Second, large scale preparation is difficult and their wide-use is unlikely.
Molecular sterics can be routinely installed onto a D-π-A scaffold, e.g., tert-butyl or ortho-substituted aryl groups (Fig. 8). The sterics may be placed onto the head group or be directly attached to the conjugative backbone. It should be noted that care must be taken so that the introduction of bulky group does not introduce internal strain. Otherwise, the fluorescence quantum yield of the dye will diminish. For a D-π-A scaffold with multiple aromatic rings, the two nearby aromatic rings can be joined together with a sp3 hybridized group.

|
Download:
|
| Fig. 8. Steric protection methods of the D-π-A scaffold. | |
{kind=link}
3. Case studies
Polymethine cyanine is a good place to get started if novel NIR dyes are sought after. Suppose a 7-atom D-π-A cyanine scaffold is what we start with (Fig. 9).

|
Download:
|
| Fig. 9. The design of polymethine cyanine dyes. | |
{kind=link}
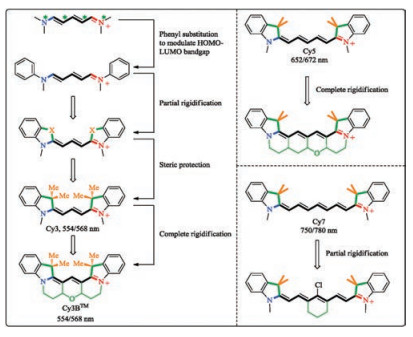
First, the electron-rich atoms of the scaffold are labeled with a star. Second, the starred atoms are judiciously substituted with electron donating groups to redshift the absorption maximum. The two phenyl groups on the two N atoms redshift the spectra by ca. 100 nm. Third, the two phenyl groups are tethered to the D-π-A scaffold by the use of "X" to inhibit the rotational freedom of the phenyl groups. Since "X" are attached to unstarred atoms of the D-π-A scaffold, an electron-donating "X", e.g., O, or N, is expected to enlarge HOMO-LUMO gap, blueshift the absorption wavelength and therefore not favorable. Instead, a sp3-hybridized carbon atom is a viable and popular choice. Fourth, onto the sp3-hybridized carbon atoms are further installed two methyl groups to yield a cyanine dye, called Cy3. The existence of the four methyl groups of Cy3 greatly reduces the tendency of the dye to aggregate. The scaffold of this Cy3 could be further rigidified leading to Cy3BTM, which exhibits a further improved fluorescence quantum yield of 0.67, compared to the 0.04 of Cy3 [121].
Both Cy3 and Cy3BTM absorbs maximally in the visible spectral regionat ca. 550 nm [122]. Since Cy3 does notlead to the desired NIR absorption, a D-π-A cyanine scaffold with a longer conjugative backbone could be employed. For example, Cy5 or Cy7 absorbs maximally in the NIR spectral region at ca. 650 nm and 750 nm, respectively (Fig. 9) [123, 124]. Due to the facile rotational freedom associated with the D-π-A backbone of Cy5 and Cy7, their fluorescence brightness is not optimal. Accordingly, a fully rigidified Cy5 was reported recently [125]. A fully rigidified Cy7 is not yet reported, presumably due to synthetic difficulty. So far, only Cy7 with a partially rigidified D-π-A backbone is available [126].
Xanthene dyes are another group of classic polymethine dyes, first reported in 1870's by von Baeye [127]. In recent years, xanthene dyes have attracted attention for imaging-based applications. Efforts have been devoted to develop long wavelength analogs of xanthenes (Fig. 10) [128-142]. Their colorconstitution relationship has been well established.

|
Download:
|
| Fig. 10. Structure-absorption relationship of xanthene dyes. | |
{kind=link}
First, enhancing the push-pull capacity of the electron donor and the acceptor can red-shift the absorption maximum, from 490 nm of fluorescein, to 585 nm of Texas red. Second, the oxygen atom bridging the two benzene rings is attached to the starred site of the xanthene skeleton. Since oxygen is electron-donating, its presence blueshifts the absorption maximum of the dye. Therefore, replacing the oxygen atom by some less electron-donating atom can redshift the absorption maximum, as clearly evidenced by the gradual shift from 548 nm of the tetramethylrhodamine, to 606 nm of carbo-rhodamine, to 643 nm of silico-rhodamine, to 700 nm of phosphor-rhodamine and further to 710 nm of sulfone-rhodamine. Third, the absorption wavelength could be redshifted by increasing the length of the conjugative backbone between the push-pull groups.
Through combined use of these structure-property relationships, we recently designed a novel NIR absorbing xanthene-type scaffold (ECX) [13], which absorbs maximally at 880 nm and emits beyond 900 nm in various solvents. Also, the NIR-absorbing/ emitting core of ECX is completely rigid and sterically protected. Therefore, ECX exhibits strong NIR fluorescence and does not aggregate. Potentials in bioimaging has been showcased. Currently, we are developing functional ECX derivatives for in vitro and in vivo translational medical applications.
4. Conclusion and outlookNIR absorbing/emitting organic dyes are sought-after to meet the ever-growing need of the biomedical imaging and theranostic applications, due to their capability for deep-tissue imaging and in vivo applications. However, most of the existing NIR absorbing dyes are not efficient emitters, as a result of fast deactivation of the excited via internal conversion and rotational freedom. Also, their poor stability is another obstacle limiting their potentials in real applications.
In this tutorial, we have summarized a general four-step approach toward development of bright NIR absorbing/emitting dyes. Following this approach, structurally succinct NIR absorbing scaffolds with good fluorescence brightness and stability could be developed.
AcknowledgmentsThe work is supported by the National Natural Science Foundation of China (Nos. 21574039 and 21822805) and Shanghai Municipal Science and Technology Commission (No. 18DZ1112703).
| [1] |
J.V. Frangioni, Curr. Opin. Chem. Biol. 7 (2003) 626-634. DOI:10.1016/j.cbpa.2003.08.007 |
| [2] |
Q.T. Nguyen, E.S. Olson, T.A. Aguilera, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 4317-4322. DOI:10.1073/pnas.0910261107 |
| [3] |
A.W. Czarnik, Chem. Biol. 2 (1995) 423-428. DOI:10.1016/1074-5521(95)90257-0 |
| [4] |
E. Haustein, P. Schwille, HFSP J. 1 (2007) 169-180. DOI:10.2976/1.2778852 |
| [5] |
J. Mei, Y. Huang, H. Tian, ACS Appl. Mater. Interfaces 10 (2018) 12217-12261. DOI:10.1021/acsami.7b14343 |
| [6] |
R. Weissleder, Science 312 (2006) 1168-1171. DOI:10.1126/science.1125949 |
| [7] |
M. Rudin, R. Weissleder, Nat. Rev. Drug Disc. 2 (2003) 123-131. DOI:10.1038/nrd1007 |
| [8] |
K. Pap, S. Plehati, I. Rajkovi c, D. Žigman, International Design Conference-Design (2010) pp. 1857-1862. |
| [9] |
BSI ISO 20473: 2007 Optics and Photonics. Spectral Bands, British Standards Institution (BSI) and International Organization for Standardisation (ISO), 2007 checked 2015.
|
| [10] |
C.M. Lin, S.M. Usama, K. Burgess, Molecules 23 (2018) 2900. DOI:10.3390/molecules23112900 |
| [11] |
M. Manley, Chem. Soc. Rev. 43 (2014) 8200-8214. DOI:10.1039/C4CS00062E |
| [12] |
M. Kamper, H. Ta, N.A. Jensen, S.W. Hell, S. Jakobs, Nat. Commun. 9 (2018) 4762. DOI:10.1038/s41467-018-07246-2 |
| [13] |
Z.H. Lei, X.R. Li, X. Luo, et al., Angew. Chem. Int. Ed. 56 (2017) 2979-2983. DOI:10.1002/anie.201612301 |
| [14] |
Z.Y. Wang, Near-Infrared Organic Materials and Emerging Applications. New York: CRC Press, 2013.
|
| [15] |
J.R. Hou, D. Jin, B. Chen, L.L. Si, Y. Li, Chin. Chem. Lett. 28 (2017) 1875-1877. DOI:10.1016/j.cclet.2017.06.017 |
| [16] |
M.S. Lin, B.A. Rasco, A.G. Cavinato, M. Al-Holy, Infrared Spectroscopy for Food Quality Analysis and Control, Elsevier Publishing, Burlington, MA, USA, 2009.
|
| [17] |
B.A. Kairdolf, A.M. Smith, T.H. Stokes, et al., Annu. Rev. Anal. Chem. 6 (2013) 143-162. DOI:10.1146/annurev-anchem-060908-155136 |
| [18] |
J. Qian, B.Z. Tang, Chemisty 3 (2017) 56-91. |
| [19] |
Z.Q. Guo, S. Park, J. Yoon, I. Shin, Chem. Soc. Rev. 43 (2014) 16-29. DOI:10.1039/C3CS60271K |
| [20] |
K.Y. Zhang, Q. Yu, H.J. Wei, et al., Chem. Rev. 118 (2018) 1770-1839. DOI:10.1021/acs.chemrev.7b00425 |
| [21] |
R. Weissleder, M.J. Pittet, Nature 452 (2008) 580-589. DOI:10.1038/nature06917 |
| [22] |
J. Zhang, R.E. Campbell, A.Y. Ting, et al., Nat. Rev. Mol. Cell Biol. 3 (2002) 906-918. DOI:10.1038/nrm976 |
| [23] |
W.R. Zipfel, R.M. Williams, W.W. Webb, Nat. Biotechnol. 21 (2003) 1369-1377. DOI:10.1038/nbt899 |
| [24] |
B. Huang, M. Bates, X.W. Zhuang, Annu. Rev. Biochem. 78 (2009) 993-1016. DOI:10.1146/annurev.biochem.77.061906.092014 |
| [25] |
V. Ntziachristos, Annu. Rev. Biomed. Eng. 8 (2006) 1-33. DOI:10.1146/annurev.bioeng.8.061505.095831 |
| [26] |
D.W. Domaille, E.L. Que, C.J. Chang, Nat. Chem. Biol. 4 (2008) 168-175. DOI:10.1038/nchembio.69 |
| [27] |
J.H. Rao, A. Dragulescu-Andrasi, H.Q. Yao, Curr. Opin. Biotechnol. 18 (2007) 17-25. DOI:10.1016/j.copbio.2007.01.003 |
| [28] |
S.L. Luo, E.L. Zhang, Y.P. Su, T.M. Cheng, C.M. Shi, Biomaterials 32 (2011) 7127-7138. DOI:10.1016/j.biomaterials.2011.06.024 |
| [29] |
E.A. Specht, E. Braselmann, A.E. Palmer, Ann. Rev. Physiol. 79 (2017) 93-117. DOI:10.1146/annurev-physiol-022516-034055 |
| [30] |
S.L. Jacques, Phys. Med. Biol. 58 (2013) R37-R61. DOI:10.1088/0031-9155/58/11/R37 |
| [31] |
L.Y. Shi, L.A. Sordillo, A. Rodríguez-Contreras, R. Alfano, J. Biophotonics 9 (2016) 38-43. DOI:10.1002/jbio.201500192 |
| [32] |
Z.P. Qin, J.C. Bischof, Chem. Soc. Rev. 41 (2012) 1191-1217. DOI:10.1039/C1CS15184C |
| [33] |
K. Welsher, Z. Liu, S.P. Sherlock, et al., Nat. Nanotechnol. 4 (2009) 773-780. DOI:10.1038/nnano.2009.294 |
| [34] |
P.Y. Wang, Y. Fan, L.F. Lu, et al., Nat. Commun. 9 (2018) 2898. DOI:10.1038/s41467-018-05113-8 |
| [35] |
J.Y. Zhao, D. Zhong, S.B. Zhou, Mater. Chem. B 6 (2018) 349-365. |
| [36] |
A.L. Antaris, H. Chen, K. Cheng, et al., Nat. Mater. 15 (2016) 235-242. DOI:10.1038/nmat4476 |
| [37] |
S. Zhu, Z. Hu, R. Tian, et al., Adv. Mater. 30 (2018) 1802546. DOI:10.1002/adma.v30.34 |
| [38] |
V.J. Pansare, S. Hejazi, W.J. Faenza, R.K. Prud'homme, Chem. Mater. 24 (2012) 812-827. DOI:10.1021/cm2028367 |
| [39] |
E. Hemmer, A. Benayas, F. Légaré, F. Vetrone, Nanoscale Horiz. 1 (2016) 168-184. DOI:10.1039/C5NH00073D |
| [40] |
J. Gao, X. Chen, Z. Cheng, Curr. Top Med. Chem. 10 (2010) 1147-1157. DOI:10.2174/156802610791384162 |
| [41] |
G. Qian, Z.Y. Wang, Chem. Asian J. 5 (2010) 1006-1029. DOI:10.1002/asia.200900596 |
| [42] |
M. Baloban, D.M. Shcherbakova, S. Pletnev, et al., Chem. Sci. 8 (2017) 4546-4557. DOI:10.1039/C7SC00855D |
| [43] |
J. Piccard, Ber. Dtsch. Chem. Ges. 46 (1913) 1843-1860. |
| [44] |
A. Mishra, R.K. Behera, P.K. Behera, B.K. Mishra, G.B. Behera, Chem. Rev. 100 (2000) 1973-2012. DOI:10.1021/cr990402t |
| [45] |
S. Daehne, Ute. Resch-Genger, O.S. Wolfbeis, Near-Infrared Dyes for High Technology Applications, Springer Science & Business Media, Berlin/Heidelberg, German (2012). |
| [46] |
J. Yao, M. Yang, Y.X. Duan, Chem. Rev. 114 (2014) 6130-6178. DOI:10.1021/cr200359p |
| [47] |
M.V. Marshall, J.C. Rasmussen, I. Tan, et al., Open Surg. Oncol. J. 2 (2010) 12-25. DOI:10.2174/1876504101002020012 |
| [48] |
I.J. Fox, E.H. Wood, Proc. Staff Meet. Mayo Clin. 35 (1960) 732-744. |
| [49] |
Z. Starosolski, R. Bhavane, K.B. Ghaghada, et al., PLoS One 12 (2017) e0187563. DOI:10.1371/journal.pone.0187563 |
| [50] |
J.T. Alander, I. Kaartinen, A. Laakso, et al., Int. J. Biomed. Imaging 2012 (2012) 940585. |
| [51] |
M.L. Landsman, G. Kwant, G.A. Mook, W.G. Zijlstra, J. Appl. Physiol. 40 (1976) 575-583. DOI:10.1152/jappl.1976.40.4.575 |
| [52] |
W. Holzer, M. Mauerer, A. Penzkofer, et al., J. Photochem. Photobiol. B Biol. 47 (1998) 155-164. DOI:10.1016/S1011-1344(98)00216-4 |
| [53] |
S. Yoneya, T. Saito, Y. Komatsu, et al., Invest. Ophthalmol. Visual Sci. 39 (1998) 1286-1290. |
| [54] |
E. Engel, T. Schraml, T. Maisch, et al., Invest. Ophthalmol. Visual Sci. 49 (2008) 1777-1783. DOI:10.1167/iovs.07-0911 |
| [55] |
D.H. Oushiki, H. Kojima, T. Terai, et al., J. Am. Chem. Soc. 132 (2010) 2795-2801. DOI:10.1021/ja910090v |
| [56] |
W. Sun, S.G. Guo, C. Hu, J.L. Fan, X.J. Peng, Chem. Rev. 116 (2016) 7768-7817. DOI:10.1021/acs.chemrev.6b00001 |
| [57] |
M.T. Sun, H. Yu, H.J. Zhu, et al., Anal. Chem. 86 (2014) 671-677. DOI:10.1021/ac403603r |
| [58] |
K. Kundu, S.F. Knight, N. Willett, et al., Angew. Chem. Int. Ed. 48 (2009) 299-303. DOI:10.1002/anie.v48:2 |
| [59] |
X.T. Jia, Q.Q. Chen, Y.F. Yang, et al., J. Am. Chem. Soc. 138 (2016) 10778-10781. DOI:10.1021/jacs.6b06398 |
| [60] |
G.T. Dempsey, M. Bates, W.E. Kowtoniuk, et al., J. Am. Chem. Soc. 131 (2009) 18192-18193. DOI:10.1021/ja904588g |
| [61] |
H. Niu, X. Jiang, J. He, et al., Tetrahedron Lett. 50 (2009) 6668-6671. DOI:10.1016/j.tetlet.2009.09.079 |
| [62] |
L. Yuan, W.Y. Lin, K.B. Zheng, L.W. He, W.M. Huang, Chem. Soc. Rev. 42 (2013) 622-661. DOI:10.1039/C2CS35313J |
| [63] |
M.Y. Li, P.C. Cui, K. Li, et al., Chin. Chem. Lett. 29 (2018) 992-994. DOI:10.1016/j.cclet.2017.11.011 |
| [64] |
Y. Yang, H. Wang, Y.L. Wei, et al., Chin. Chem. Lett. 28 (2017) 2023-2026. DOI:10.1016/j.cclet.2017.08.051 |
| [65] |
S. Dähne, Science 199 (1978) 1163-1167. DOI:10.1126/science.199.4334.1163 |
| [66] |
J. Griffiths, Colour and Constitution of Organic Molecules. London: Academic Press, 1976.
|
| [67] |
R.L.M. Allen, Colour Chemistry. 1st ed.. New York: Appleton-Century-Crofts, 1971.
|
| [68] |
J. Clayden, N. Greeves, S. Warren, Organic Chemistry. 2nd ed.. , Oxford University Press, 2001.
|
| [69] |
R.L. Christensen, A. Faksh, J.A. Meyers, et al., J. Phys. Chem. A 108 (2004) 8229-8236. DOI:10.1021/jp048421g |
| [70] |
J. Rissler, Chem. Phys. Lett. 395 (2004) 92-96. DOI:10.1016/j.cplett.2004.07.058 |
| [71] |
C.B. Gorman, E.J. Ginsburg, S.R. Marder, R.H. Grubbs, Angew. Chem. Int. Ed. 28 (1989) 1571-1574. |
| [72] |
K. Mullen, G. Wegner, Electronic Materials: The Oligomer Approach, WileyVCH, Berlin, 1998.
|
| [73] |
R. Duval, C. Duplaisb, Nat. Prod. Rep. 34 (2017) 161-193. DOI:10.1039/C6NP00111D |
| [74] |
W.P. Chen, R.F. Chen, Q.P. Liu, Y. He, K. He, Chem. Sci. 8 (2017) 4917-4925. DOI:10.1039/C7SC00475C |
| [75] |
J.M. Gao, S.X. Yang, J.C. Qin, Chem. Rev. 113 (2013) 4755-4811. DOI:10.1021/cr300402y |
| [76] |
J. Bendig, U. Schedler, T. Harder, et al., J. Photochem. Photobiol. A 91 (1995) 53-57. DOI:10.1016/1010-6030(95)04108-R |
| [77] |
S.A. Tikhonov, V.I. Vovna, N.A. Gelfand, et al., J. Phys. Chem. A 120 (2016) 7361-7369. DOI:10.1021/acs.jpca.6b07242 |
| [78] |
A. Chaudhuri, Y. Venkatesh, K.K. Behara, N.D.P. Singh, Org. Lett. 19 (2017) 1598-1601. DOI:10.1021/acs.orglett.7b00416 |
| [79] |
S. Imazeki, A. Mukoh, T. Yoneyama, M. Kaneko, Mol. Cryst. Liq. Cryst. 145 (1987) 79-93. DOI:10.1080/00268948708080215 |
| [80] |
Y.M. Shen, Z.H. Shang, Y.H. Yang, et al., J. Org. Chem. 80 (2015) 5906-5911. DOI:10.1021/acs.joc.5b00242 |
| [81] |
Y. Xiao, F.Y. Liu, Z. Chen, et al., Chem. Commun. 51 (2015) 6480-6488. DOI:10.1039/C4CC09846C |
| [82] |
A. Treibs, K. Jacob, Justus Liebigs Ann. Chem. 699 (1966) 153-167. |
| [83] |
A. Schmitt, B. Hinkeldey, M. Wild, G. Jung, J. Fluoresc. 19 (2009) 755-758. DOI:10.1007/s10895-008-0446-7 |
| [84] |
M.Y. Fu, Y. Xiao, X.H. Qian, D.F. Zhao, Y.F. Xu, Chem. Commun. 15 (2008) 1780-1782. |
| [85] |
G. Sathyamoorthi, M.L. Soong, T.W. Ross, J.H. Boyer, Heteroat. Chem. 4 (1993) 603-608. |
| [86] |
A.K.-Y. Jen, Y.Q. Liu, L.X. Zheng, et al., Adv. Mater. 11 (1999) 452-455. |
| [87] |
Y.Y. Cheng, G.C. Li, Y. Liu, et al., J. Am. Chem. Soc. 138 (2016) 4730-4738. DOI:10.1021/jacs.5b09241 |
| [88] |
W.M. Liu, B.J. Zhou, G. Niu, et al., ACS Appl. Mater. Interfaces 7 (2015) 7421-7427. DOI:10.1021/acsami.5b01429 |
| [89] |
L. Yuan, W.Y. Lin, Y.T. Yang, H. Chen, J. Am. Chem. Soc. 134 (2012) 1200-1211. DOI:10.1021/ja209292b |
| [90] |
M. Sibrian-Vazquez, J.O. Escobedo, M. Lowry, R.M. Strongin, Pure Appl. Chem. 84 (2012) 2443-2456. DOI:10.1351/PAC-CON-11-11-06 |
| [91] |
E. Azuma, N. Nakamura, K. Kuramochi, et al., J. Org. Chem. 77 (2012) 3492-3500. DOI:10.1021/jo300177b |
| [92] |
A.B. Descalzo, K. Rurack, Chem. Eur. J. 15 (2009) 3173-3185. DOI:10.1002/chem.v15:13 |
| [93] |
R.S. Lepkowicz, C.M. Cirloganu, O.V. Przhonska, et al., Chem. Phys. 306 (2004) 171-183. DOI:10.1016/j.chemphys.2004.07.021 |
| [94] |
A. Treibs, K. Jacob, Angew. Chem. Int. Ed. 4 (1965) 694-694. |
| [95] |
X.Z. Song, J.W. Foley, Dyes Pigm. 78 (2008) 60-64. DOI:10.1016/j.dyepig.2007.10.006 |
| [96] |
B.H. Li, L.F. Lu, M.Y. Zhao, Z.H. Lei, F. Zhang, Angew. Chem. Int. Ed. 57 (2018) 7483-7487. DOI:10.1002/anie.201801226 |
| [97] |
B.L. Guennic, D. Jacquemin, Acc. Chem. Res. 48 (2015) 530-537. DOI:10.1021/ar500447q |
| [98] |
K.Z. Gu, Y.S. Xu, H. Li, et al., J. Am. Chem. Soc. 138 (2016) 5334-5340. DOI:10.1021/jacs.6b01705 |
| [99] |
N. Karton-Lifshin, L. Albertazzi, M. Bendikov, et al., J. Am. Chem. Soc. 134 (2012) 20412-20420. DOI:10.1021/ja308124q |
| [100] |
H.Y. Li, X.H. Li, W. Shi, Y.H. Xu, H.M. Ma, Angew. Chem. Int. Ed. 57 (2018) 12830-12834. DOI:10.1002/anie.201808400 |
| [101] |
L. Strekowski, Heterocyclic Polymethine Dyes: Synthesis, Properties and Applications, Springer-Verlag, Berlin, 2008.
|
| [102] |
K. Umezawa, Y. Nakamura, H. Makino, D. Citterio, K. Suzuki, J. Am. Chem. Soc. 130 (2008) 1550-1551. DOI:10.1021/ja077756j |
| [103] |
M. Irie, Chem. Rev. 100 (2000) 1685-1716. DOI:10.1021/cr980069d |
| [104] |
G.L. Niu, W.M. Liu, B.J. Zhou, et al., J. Org. Chem. 81 (2016) 7393-7399. DOI:10.1021/acs.joc.6b00981 |
| [105] |
D. Wu, Y.Z. Shen, J.H. Chen, et al., Chin. Chem. Lett. 28 (2017) 1979-1982. DOI:10.1016/j.cclet.2017.07.004 |
| [106] |
F.L. Song, R. Liang, J.D. Deng, Z.W. Liu, X.J. Peng, Chin. Chem. Lett. 28 (2017) 1997-2000. DOI:10.1016/j.cclet.2017.08.023 |
| [107] |
Z.H. Lei, Z.H. Zeng, X.H. Qian, Y.J. Yang, Chin. Chem. Lett. 28 (2017) 2001-2004. DOI:10.1016/j.cclet.2017.09.023 |
| [108] |
P. Ning, W.J. Wang, M. Chen, Y. Feng, X.M. Meng 28 (2017) 1943-1951. |
| [109] |
X.T. Jia, Q.Q. Chen, Y.F. Yang, et al., J. Am. Chem. Soc. 138 (2016) 10778-10781. DOI:10.1021/jacs.6b06398 |
| [110] |
A.P. Gorka, R.R. Nani, J.J. Zhu, S. Mackem, M.J. Schnermann, J. Am. Chem. Soc. 136 (2014) 14153-14159. DOI:10.1021/ja5065203 |
| [111] |
X. Jing, C. He, L. Zhao, C.Y. Duan, Acc. Chem. Res. 52 (2019) 100-109. DOI:10.1021/acs.accounts.8b00463 |
| [112] |
S.Y. Hsueh, C.C. Lai, Y.H. Liu, et al., Org. Lett. 9 (2007) 4523-4526. DOI:10.1021/ol702050w |
| [113] |
A.G. Cheetham, T.D.W. Claridge, H.L. Anderson, Org. Biomol. Chem. 5 (2007) 457-462. DOI:10.1039/b616621k |
| [114] |
E.J.F. Klotz, T.D.W. Claridge, H.L. Anderson, J. Am. Chem. Soc. 128 (2006) 15374-15375. DOI:10.1021/ja0665139 |
| [115] |
E. Arunkumar, C.C. Forbes, B.C. Noll, B.D. Smith, J. Am. Chem. Soc. 127 (2005) 3288-3289. DOI:10.1021/ja042404n |
| [116] |
Z.M. Tao, G.S. Hong, C. Shinji, et al., Angew. Chem. Int. Ed. 125 (2013) 13240-13244. DOI:10.1002/ange.201307346 |
| [117] |
S. Hecht, J.M.J. Frechet, Angew. Chem. Int. Ed. 40 (2001) 74-91. DOI:10.1002/1521-3773(20010105)40:1<>1.0.CO;2-6 |
| [118] |
K. Yanagi, K. Iakoubovskii, H. Matsui, et al., J. Am. Chem. Soc. 129 (2007) 4992-4997. DOI:10.1021/ja067351j |
| [119] |
X.M. Wu, S. Chang, X.R. Sun, Chem. Sci. 4 (2013) 1221-1228. DOI:10.1039/c2sc22035k |
| [120] |
J.E.H. Buston, J.R. Young, H.L. Anderson, Chem. Commun. 11 (2000) 905-906. |
| [121] |
M. Cooper, A. Ebner, M. Briggs, et al., J. Fluoresc. 14 (2004) 145-150. DOI:10.1023/B:JOFL.0000016286.62641.59 |
| [122] |
C.Y. Fan, J.C. Hsiang, R.M. Dickson, ChemPhysChem 13 (2012) 1023-1029. DOI:10.1002/cphc.201100671 |
| [123] |
B. Li, Z. He, H. Zhou, H. Zhang, T. Cheng, Chin. Chem. Lett. 28 (2017) 1929-1934. DOI:10.1016/j.cclet.2017.08.055 |
| [124] |
J.L. Bricks, A.D. Kachkovskii, Y.L. Slominskii, A.O. Gerasov, S.V. Popov, Dyes Pigm. 121 (2015) 238-255. DOI:10.1016/j.dyepig.2015.05.016 |
| [125] |
M.S. Michie, R. Götz, C. Franke, et al., J. Am. Chem. Soc. 139 (2017) 12406-12409. DOI:10.1021/jacs.7b07272 |
| [126] |
M.L. Korb, Y.E. Hartman, J. Kovar, et al., J. Surg. Res. 188 (2014) 119-128. DOI:10.1016/j.jss.2013.11.1089 |
| [127] |
A. Baeyer, Ber. Dtsch. Chem. Ges. 4 (1871) 555-558. |
| [128] |
Y.J. Yang, M. Lowry, X.Y. Xu, et al., Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 8829-8834. DOI:10.1073/pnas.0710341105 |
| [129] |
J.B. Grimm, A.J. Sung, W.R. Legant, et al., ACS Chem. Biol. 8 (2013) 1303-1310. DOI:10.1021/cb4000822 |
| [130] |
T. Egawa, Y. Koide, K. Hanaoka, et al., Chem. Commun. 47 (2011) 4162-4164. DOI:10.1039/c1cc00078k |
| [131] |
A. Fukazawa, S. Suda, M. Taki, et al., Chem. Commun. 52 (2016) 1120-1123. DOI:10.1039/C5CC09345G |
| [132] |
J.B. Grimm, B.P. English, J.J. Chen, et al., Nat. Methods 12 (2015) 244-250. DOI:10.1038/nmeth.3256 |
| [133] |
L. Wu, K. Burgess, Org. Lett. 10 (2008) 1779-1782. DOI:10.1021/ol800526s |
| [134] |
Y. Koide, Y. Urano, K. Hanaoka, T. Terai, T. Nagano, ACS Chem. Biol. 6 (2011) 600-608. DOI:10.1021/cb1002416 |
| [135] |
C. Aaron, C.C. Barker, J. Chem. Soc. (1986) 2655-2662. |
| [136] |
X.Q. Zhou, R. Lai, J.R. Beck, H. Li, C.I. Stains, Chem. Commun. 52 (2016) 12290-12293. DOI:10.1039/C6CC05717A |
| [137] |
J. Liu, Y.Q. Sun, H.X. Zhang, et al., ACS Appl. Mater. Interfaces 8 (2016) 22953-22962. DOI:10.1021/acsami.6b08338 |
| [138] |
M. Sauer, K.T. Han, R. Muller, et al., J. Fluoresc. 5 (1995) 247-261. DOI:10.1007/BF00723896 |
| [139] |
M. Sauer, K.T. Han, R. Muller, et al., J. Fluoresc. 3 (1993) 131-139. DOI:10.1007/BF00862730 |
| [140] |
O.O. Abugo, R. Nair, J.R. Lakowicz, Anal. Biochem. 279 (2000) 142-150. DOI:10.1006/abio.2000.4486 |
| [141] |
A.V. Anzalone, T.Y. Wang, Z.X. Chen, V.W. Cornish, Angew. Chem. Int. Ed. 52 (2013) 650-654. DOI:10.1002/anie.201205369 |
| [142] |
J. Arden-Jacob, J. Frantzeskos, N.U. Kemnitzer, A. Zilles, K.H. Drexhage, Spectrochimica Acta Part A 57 (2001) 2271-2283. DOI:10.1016/S1386-1425(01)00476-0 |