 2019, Vol. 30
2019, Vol. 30 


b Beijing Key Lab of Green Recycling and Extraction of Rare and Precious Metals, University of Science and Technology Beijing, Beijing 100083, China
Silver nanoparticles (Ag NPs) have attracted a lot of attentions in electronic, catalytic, sensing, and biological applications in the past decades due to their strong size/shape-dependent physiochemical properties and good oxidation resistance [1-5]. The controllable preparation of Ag NPs on large scale was urgently demanded to meet the requirements of above industries. Many processes have been invented to prepare the Ag NPs, including evaporation/condensation [6, 7], photo-induced transformation [8-10], spray pyrolysis [11-13], and liquid phase reductive process (LPRP) [14-27] and so on, among which the LPRP has been widely applied because of its moderate conditions and facile manipulation [14, 17, 18]. Generally in order to guarantee Ag NPs with regular size and shape by using LPRP, low concentration of free Ag+ and low reduction speed must be created for homogenous nucleation [14, 17, 23], resulting in the difficulty of large-scale synthesis of Ag NPs. In previous reports, adding some anions (Cl-, Br-, I-, CO32-, C2O42-, SO32-, SCN-) to form insoluble sliver salts has been confirmed to be one of the most effective strategies to control the size and morphology of Ag NPs, decrease the concentration of free Ag+, avoid the second nucleation and improve the yield of the product [17-26]. While, few reports systematically compared the characteristics of insoluble silver salts with their corresponding reduction products. Chen and coworkers [17] have prepared synthesized size-controllable sliver nanoplates by reducing silver nitrate in the presence of different insoluble silver salts; Tao and Xia [20] have produced monodisperse and quasi-spherical Ag NPs in water via adding I-, Br- and Cl-, but the size of obtained products is not very different in their synthetic system respectively. Interestingly in our work, the silver halide particles with different sizes of 110 nm AgCl, 60 nm AgBr, and 30 nm AgI were reproducibly prepared on large scale by controlled double-jet precipitation (CDJP) method [28-30], and then were further used for reduction by ascorbic acid or sodium borohydride. And it is found that the size of obtained Ag NPs was strongly dependent on the AgX size under the same synthesis conditions. Therefore, the present study will take an exploration on this system from the viewpoint of size-dependent relationship between the AgX and Ag NPs.
The first step in our work is the synthesis of colloidal AgX products. In a typical synthetic approach, 500 mL of deionized water containing 10 g PVP was introduced into a 2500 mL beaker. Then, a solution containing AgNO3 (0.1 mol/L) and a solution containing NaX (0.11 mol/L) were dropped into the reactor in a simultaneous way at the flow rate of 5 mL/min by peristaltic pumps respectively. Agitation at 250 rpm was set to disperse the solution and particles using a magnetic stirrer in a temperaturecontrolled water bath at 25 ℃. The total injection time was adjusted to 100 min, after dropping, the suspension was kept aging for 30 min under the stirring state. Particularly, above operations were carried out in dark conditions to avoid photodecomposition effect. For further characterization, the as-prepared AgX were separated by centrifugation and washed with Milli-Q water and ethanol respectively for three times.
Then different reducing agents (ascorbic acid and sodium borohydride) are used to reduce AgX to prepare the Ag particles. The typical steps for preparing Ag particles with ascorbic acid are as follows. A certain amount of ascorbic acid was placed into above AgX suspensions (500 mL, pH 3.0) quickly, stirred intensively 5 min to approach homogeneous state, and the molar ratio of ascorbic acid to AgX was 3:2 to ensure the complete reduction of silver halides. After that, above suspensions were fed with 6 mol/L NaOH solution to adjust the pH to a set value. Then these mixtures were stirred for different period of time at a certain temperature. Similarly, all procedures required dark conditions due to the sensitivity of AgX to light. A series of Ag nanoparticles with different sizes were obtained by choosing the different silver halides and reductive reagents as well as the reaction conditions, and the products were collected by centrifugation and washed by water. The typical steps for preparing Ag particles with sodium borohydride are as follows. Ten milliliter 0.5 mol/L and 2 mol/L sodium borohydride solution was dipped into the 50 mL AgX suspensions respectively at a feeding rate of 2 mL/min, and then the obtained suspensions were put in dark conditions for aging 2 h. The products were collected by centrifugation and washed by water.
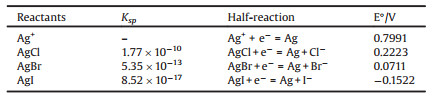
Fig. 1 indicated that the obtained silver halide particles were well dispersed and had the clear average size order as following: 110 nm AgCl > 60 nm AgBr > 30 nm AgI, and the X-ray diffraction patterns also verified the pure AgX phases obtained in respective solution system, and no metallic Ag was generated in this process. Because the double-jet precipitation process was performed in the same set-ups under the same operational parameters, it could be ascribed this variation trend to the crystallization habit of each AgX. Due to the Ksp constants of AgCl, AgBr and AgI were 1.8×10-10, 5.0×10-13, and 8.3×10-17, respectively, so when the two reagents were mixed in a double-jet feeding way, the preferential nucleation would have the following order: AgI > AgBr > AgCl. According to the empirical law of Weimarn [31, 32], it was easy to predict that the nucleation rate of AgI would become the largest that led to the finest precipitated particles. As demonstrated in Fig. 1, it could be confirmed that it was the difference in the dissolubility of AgCl, AgBr and AgI that led to the different particle size. It also could be seen that the bottle samples in Fig. 1 had the quite different colors and transparent degree, suggesting that the precipitated AgX particles were quite fine and had the strong sizedependent optical properties.

|
Download:
|
| Fig. 1. The images of AgX nanoparticles obtained by CDJP technique: (a) SEM, AgCl, (c) SEM, AgBr and (e) TEM, AgI; XRD patterns of AgX crystals: (b) AgCl, (d) AgBr, (f) AgI. | |
As demonstrated in Fig. 2, compared with AgX, the morphology of Ag NPs obtained by reduction of ascorbic acid changed clearly, meanwhile the XRD of the products were metallic Ag with a facecentered cubic structure (Fig. 3), indicating that all the AgX particles were transferred into Ag under these synthetic systems. During the reducing silver halide to silver powder, reaction time and solution pH have drastic effect on particle purity, shape and size, which are very similar to the results of other research works [18, 23, 25]. At pH 12, the obtained Ag particles by reduction with ascorbic acid from the AgCl had a larger size and irregular shapes, while monodispersed Ag spherical particles with around 70nm size were obtained from AgBr in this case, and much finer spherical Ag NPs from AgI with the average size of 30nm. TEM micrographs verified that the Ag NPs reduced from the AgBr and AgI were quite dispersed and smooth in surface. In order to prepare the monodispersed Ag NPs from the AgCl, it was interesting to find that by adjusting the solution pH down to weak acidic condition, e.g., 5.0 and heating at 60 ℃, the obtained reductive Ag particles had the mean size of around 240nm with quite nice dispersity and uniformity, which was related to the reduction activity of ascorbic acid at different solution pH values [23]. In a word, the final products of Ag NPs had the strong size-dependent relationship with the AgX sizes, and were positively influenced by the latter. Above discussionshowedus a clear size diminishing trendwith the decreasing of Ksp(AgX) constants. In order to learn more about the Ag NPs size variation trend and the relative mechanisms, the reduction potential values of AgX/Ag(0) were listed inTable 1, and it could be found that AgCl was the easiest sample to be reduced into Ag particles among the three AgX, and it also suggested that the direct reduction of Ag+ into Ag(0) was quite fast and it could be assumed that the obtained silver particles were hard to have the good dispersity and morphology due to too fast reaction. Actually, this empirical law could find the proof in the case of reduction of AgCl at pH 12, and the reduction rate of AgX to Ag was strongly influenced by pH, i.e., higher the pH, faster the reduction of AgX intoAg. So for the different AgX, we set the different pH and time in the reduction experiments, as shown in Fig. 2. Otherwise, the optimal reduction conditions for AgCl would not reduce the AgBr and AgI colloids, and even could not obtain any silver metallic particles.

|
Download:
|
| Fig. 2. Images of Ag particles obtained by reducing AgX with ascorbic acid under the following conditions: (a, SEM) AgCl, pH 12, 0.5 h, 25 ℃; (b, SEM) AgCl, pH 5, 1 h, 60 ℃; (c, SEM; d, TEM) AgBr, pH 12, 2 h, 25 ℃; (e, SEM; f, TEM) AgI, pH 12, 8 h, 25 ℃. | |

|
Download:
|
| Fig. 3. XRD patterns of the obtained silver particles by reducing AgX with ascorbic acid under the following conditions: (a) AgCl, pH 5, 1h, 60 ℃; (b) AgBr, pH 12, 2h, 25 ℃; (c) AgI, pH 12, 8h, 25 ℃. | |
Fig. 4 indicated the effect of concentration of sodium borohydride as the reductant on the size and morphology of the obtained Ag NPs, and it could be seen that from the AgCl samples, Ag particles were seriously aggregated and of irregular shapes. But the Ag NPs obtained from the AgBr and AgI particles were well dispersed and uniform in size (Figs. 4c and d), with about 35nm and 20nm average size respectively. It also could be found that high concentration of the sodium borohydride solution would make the size distribution of Ag NPs broader with much finer tiny particles. In general, strong reducing agents are used to prepare small Ag NPs, the use of weak reductants results in the formation of large Ag NPs [21]. By comparing with the Ag NPs samples obtained by reduction of ascorbic acid, it could be found that the silver nanoparticles would become much smaller in the case of sodium borohydride as the reductant, which was also easy to understand that the much larger nucleation rate occurred in the case of much stronger reduction capability of sodium borohydride.

|
Download:
|
| Fig. 4. Photographs of the silver particles obtained by reduction the AgX (a: AgCl; c: AgBr; e: AgI) by 10mL 0.5mol/L sodium borohydride at a feeding rate of 2mL/min, and by 10mL 2mol/L sodium borohydride at a feeding rate of 2mL/min for AgCl (b), AgBr (d), AgI (f). | |
|
|
Table 1 Basic constants of the AgX/Ag solution systems. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
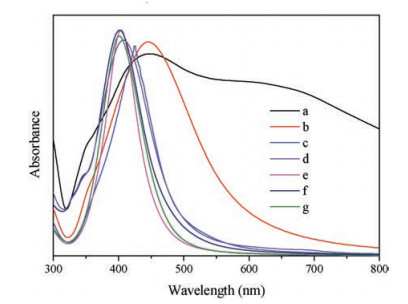
Inorder to learnmoreabout the size-dependent optical properties, the UV spectrometric measurements were conducted, and the results were shown in Fig. 5. It was found that the Ag NPs obtained from different AgX particles have different particle size with good dispersity, so the test spectra would reflect the correspondingsize-relatedoptical properties. It was shown that the remarkable blue shift towards shorter wavenumbers of the characteristic peaks occurred with the decreasing of particle size of Ag NPs, which was also verified in many other research works [21, 33].

|
Download:
|
| Fig. 5. UV spectra of the Ag NPs under the following conditions: curves a–g corresponding to Figs. 2b–d, Figs. 2e and f, Figs. 4c–f, respectively. | |
{kind=link}
Monodispersed silver nanoparticles were prepared by using AgX colloids obtained by controlled double-jet precipitation and further reduced into Ag NPs by reductants of ascorbic acid or sodium borohydride. The obtained AgX particle size affected the Ag NPs size greatly, and they had the positive relationship in the size variation. Smaller the AgX particles, finer the Ag NPs particles and this phenomenon could be well explained by the relative ratios of the nucleation rate to the growth one under various conditions. Blue shift occurs with the size decreasing of the prepared monodispersed Ag NPs, which gave a good example to show the confirmed size-dependent relationship with the optical properties. The present synthesis process has the prominent advantage of more controllability for the whole process, which is also good for the scaling-up due to the avoidance of fast reduction process.
| [1] |
H. You, J. Fang, Nano Today 11 (2016) 145-167. DOI:10.1016/j.nantod.2016.04.003 |
| [2] |
H. Liu, T. Liu, L. Zhang, et al., Adv. Funct. Mater. 25 (2015) 5435-5443. DOI:10.1002/adfm.201502366 |
| [3] |
L. Qin, G. Zeng, C. Lai, et al., Sensor. Actuat. B:Chem. 243 (2017) 946-954. DOI:10.1016/j.snb.2016.12.086 |
| [4] |
K. Zheng, M.I. Setyawati, D.T. Leong, J. Xie, Coord. Chem. Rev. 357 (2018) 1-17. DOI:10.1016/j.ccr.2017.11.019 |
| [5] |
X. Jiang, B. Du, Y. Huang, J. Zheng, Nano Today 21 (2018) 106-125. DOI:10.1016/j.nantod.2018.06.006 |
| [6] |
N.K. Manninen, N.M. Figueiredo, S. Carvalho, A. Cavaleiro, Plasma Process Polym. 11 (2014) 629-638. DOI:10.1002/ppap.201300175 |
| [7] |
N.M. Figueiredo, R. Serra, N.K. Manninen, A. Cavaleiro, Appl. Surf. Sci. 440 (2018) 144-152. DOI:10.1016/j.apsusc.2018.01.011 |
| [8] |
H. Yu, Q. Zhang, H. Liu, et al., ACS Nano 8 (2014) 10252-10261. DOI:10.1021/nn503459q |
| [9] |
R. Jin, Y. Cao, C.A. Mirkin, et al., Science 294 (2001) 1901-1903. DOI:10.1126/science.1066541 |
| [10] |
Q. Zhang, N. Li, J. Goebl, Z. Lu, Y. Yin, J. Am. Chem. Soc. 133 (2011) 18931-18939. DOI:10.1021/ja2080345 |
| [11] |
D.S. Jung, H.Y. Koo, Y.C. Kang, J. Colloid Interfaces Sci. 343 (2010) 1-6. DOI:10.1016/j.jcis.2009.05.070 |
| [12] |
N. Kumar, F. Alam, V. Dutta, J. Alloys Compd. 585 (2014) 312-317. DOI:10.1016/j.jallcom.2013.09.145 |
| [13] |
X. Shi, S. Wang, X. Duan, Q. Zhang, Mater. Chem. Phys. 112 (2008) 1110-1113. DOI:10.1016/j.matchemphys.2008.07.043 |
| [14] |
X.W. Han, X.F. Zeng, J. Zhang, et al., Chem. Eng. J. 296 (2016) 182-190. DOI:10.1016/j.cej.2016.03.076 |
| [15] |
X. Wang, S. Xu, W. Xu, Nanoscale 3 (2011) 4670-4675. DOI:10.1039/c1nr10590f |
| [16] |
B. Khodashenas, H.R. Ghorbani, Arab. J. Chem. (2015), doi: http://dx.doi.org/10.1016/j.arabjc.2014.12.014.
|
| [17] |
X. Zhao, B. Chen, C. Li, et al., Chem. Eng. J. 285 (2016) 690-697. DOI:10.1016/j.cej.2015.10.041 |
| [18] |
M. Liu, M. Leng, C. Yu, X. Wang, C. Wang, Nano Res. 3 (2010) 843-851. DOI:10.1007/s12274-010-0055-z |
| [19] |
Z. Wang, J. Liu, X. Chen, J. Wan, Y. Qian, J. Am. Chem. Soc. 11 (2004) 160-163. |
| [20] |
H. Li, H. Xia, D. Wang, X. Tao, Langmuir 29 (2013) 5074-5079. DOI:10.1021/la400214x |
| [21] |
Y. Cao, R. Zheng, X. Ji, et al., Langmuir 30 (2014) 3876-3882. DOI:10.1021/la500117b |
| [22] |
S. Liu, J. Yue, A. Gedanken, Adv. Mater. 13 (2001) 656-658. |
| [23] |
B. Chen, X. Jiao, D. Chen, Cryst. Growth Des. 10 (2010) 3378-3386. DOI:10.1021/cg901497p |
| [24] |
Z. Wang, J. Liu, X. Chen, J. Wan, Y. Qian, Chem. -Eur. J. 11 (2005) 160-163. |
| [25] |
S. Peng, Y. Sun, Chem. Mater. 22 (2010) 6272-6279. DOI:10.1021/cm101814f |
| [26] |
N. Cathcart, N. Coombs, I. Gourevich, V. Kitaev, Nanoscale 8 (2016) 18282-18290. DOI:10.1039/C6NR07397B |
| [27] |
Y.P. Zhu, X.K. Wang, W.L. Guo, J.G. Wang, C. Wang, Ultrason. Sonochem. 17 (2010) 675-679. DOI:10.1016/j.ultsonch.2010.01.003 |
| [28] |
S.H. Lee, Y.S. Her, E. Matijevic, J. Colloids Interfaces Sci. 186 (1997) 193-202. DOI:10.1006/jcis.1996.4638 |
| [29] |
A. Safronikhin, H. Ehrlich, G. Lisichkin, J. Alloys Compd. 694 (2017) 1182-1188. DOI:10.1016/j.jallcom.2016.10.128 |
| [30] |
X. Yan, L. Chai, Q. Li, et al., CrystEngComm 18 (2016) 924-929. DOI:10.1039/C5CE01916H |
| [31] |
F.P. Ludwig, J. Schmelzer, J. Colloid Interface Sci. 181 (1996) 503-510. DOI:10.1006/jcis.1996.0407 |
| [32] |
P.P.V. Weimarn, Chem. Rev. 2 (2002) 217-242. |
| [33] |
X. Liu, L. Li, Y. Yang, Y. Yin, C. Gao, Nanoscale 6 (2014) 4513-4516. DOI:10.1039/c4nr00254g |