 2019, Vol. 30
2019, Vol. 30 


b Research Institute for Energy Equipment Materials, Hebei University of Technology, Tianjin 300130, China;
c Tianjin Key Laboratory of Laminating Fabrication & Interface Control Technology for Advanced Materials, Tianji 300130, China
Nasicon based cathode materials have recently been paid much attention due to their elasticthree-dimensional(3D)framework that accommodates compositional changes and provides a fast pathway for ion transport [1-10]. Among them, Li3Fe2(PO4)3 is widely investigated from both economic and environmental viewpoints [3-11]. It presents a theoretical capacity of 128 mAh/g vs. Fe2+/Fe3+ redox couple. However, poor electronic conductivity (< 10-8 S/cm) caused by too large separation between the metal ions significantly retards its practical capacity. By decreasing the dimensions of the active material particles and introducing the conductive carbon to improve the interfacial connectivity, Morcrette et al. [11] obtained a high reversible capacity of 110 mAh/gata rate of 0.05C and about 85 mAh/g at 0.5C for the compound. Unfortunately, it is difficult to meet the rapid growing demand of high energy/power densities batteries when the discharging rate is lower than 0.5C. Therefore, great efforts are still needed to optimize electrochemical performance of the Li3Fe2(PO4)3 compound.
Transition metal ion substitution was generally adopted to improve electrochemical properties of the Li3Fe2(PO4)3 [12-16]. Sun et al. [15] found that the Ti4+ and Mn2+ doped Li3Fe1.8- Mn0.1Ti0.1(PO4)3 presented a high discharging capacity of 94 mAh/g at 0.5C rate. Furthermore, Liu et al. [16] proved that single Ti4+-substituded Li2.8Fe2Ti0.2(PO4)3 showed better electrochemical properties than the abovementioned Ti4+ and Mn2+-substituted one. Its initial discharging capacity was about 122.3 mAh/g at 0.05C rate, which is very close to the theoretical capacity of Li3Fe2(PO4)3. And a high discharging capacity of about 100 mAh/g was obtained at 0.5C rate after 20 charging-discharging cycles for the Ti4+-substituted Li2.8Fe2Ti0.2(PO4)3 compound.
In spite of few reports on anionic substitution for the Nasicon cathode materials [17], we recently used several polyanions such as VO43- [18], MoO42- [19] and SO42- [20] to substitute for partial PO43- in the Li3Fe2(PO4)3, based on the successful achievements of the mixed anion effect on solid state electrolytes [21-26]. It reveals that all these polyanion substituted products exhibited the initial discharging capacity larger than 95 mAh/g, with more than 94% capacity retention after 60 charge-discharge cycles at 2C rate, showing superior performances than those of the abovementioned transition metal ion doped ones.
Simultaneous substitution of cation and anion was initially used to enhance ionic conductivity of the Ag+ and Cu+ superion conductors like Ag4HgSe2I2 [27] and Rb4Cu16Cl13I7 [28], this measure was proved to be also effective on LiTi2(PO4)3 solid state electrolyte [29, 30]. Based on the abovementioned beneficial effects caused by cations and/or anions substitution, we were motivated to introduce Ti4+ along with VO43- to the Li3Fe2(PO4)3 cathode material, aiming to boost its electrochemical performance. This paper deals with substitution of Ti4+ for the Fe3+ in our formerly reported Li3Fe2(PO4)2.55(VO4)0.45 cathode material. Effect of simultaneous substitution of Ti4+ and V5+ on electrochemical properties was investigated for the Li3Fe2(PO4)3 cathode material.
Reagent-grade LiNO3 (purity 99%), Fe(NO3)3·9H2O (purity 98.5%), NH4H2PO4 (purity 99%), Li2CO3 (purity 98%), V2O5 (purity 99%) and Ti(C4H9O)4 (purity 99%) were used as starting materials for synthesis of the nominal Li3-xTixFe2-x(PO4)2.55(VO4)0.45 (x = 0~0.3) compound. The starting materials LiNO3, Fe(NO3)3·9H2O and NH4H2PO4 were dissolved in a stoichiometric ratio in 200 dm3 DI water to achieve the total metal ions concentration of 0.06 mol/L. Further, V2O5 and Li2CO3 were dissolved in a stoichiometric ratio of Li3VO4 in 0.21 mol/L HNO3 solution, maintaining the VO43- concentration of 0.1 mol/L [31]. Those two prepared solutions were mixed, and the same amount of citric acid and glycol was added and dissolved in the resulting solution as the complexing agent and dispersant, respectively, with the molar ratio of citric acid to metal ions equals to 1:1. On the other hand, appropriate Ti(C4H9O)4 was dissolved into alcohol to form a homogeneous solution, which was then added to the above mixed solution. The formed solution was thoroughly stirred, by adding ammonia (30%) to adjust the pH value at about 5.0. The final solution was heated at 80 ℃ to evaporate the excess water and formed a gel. After keeping for 48 h, the gel was calcined at 600 ℃ to obtain the final products.
Material characterization and electrochemical measurements were described in our previous work [18].
Fig. 1a shows XRD patterns of the nominal Li3-xTix Fe2-x(PO4)2.55(VO4)0.45 (x = 0~0.3) samples heated at 600 ℃. By contrast, the standard XRD patterns of Li3Fe2(PO4)3, V2Ti7O17 and Fe2O3 are also presented in this figure. The single VO43- substituted sample, i.e., x = 0 or Li3Fe2(PO4)2.55(PO4)0.45, was identified as the pure Li3Fe2(PO4)3 phase with an ordered monoclinic structure, corresponding to the JCPDS No. 80-1515. Introduction of Ti4+ cations into the Li3Fe2(PO4)2.55(VO4)0.45 results in some identified impurity peaks located at 2θ of around 29°, 22° and 35° in addition to the Li3Fe2(PO4)3 phase, which may correspond to the phases like V2Ti7O17 and Fe2O3, respectively. Those results suggest that, even though a pure Li3Fe2(PO4)3 phase could be prepared by the Ti4+- substituted samples, along with the VO43- doping, a little impure phases were easy to appear in the obtained products.

|
Download:
|
| Fig. 1. XRD patterns of the samples in a nominal composition of Li3-xTixFe2-x(PO4)2.55(VO4)0.45 heated at 600 ℃, (a) large survey XRD patterns, (b) deconvolution to the peaks at 2θ of about 20°–22°. | |
{kind=link}
It is well established that two stronger XRD peaks, namely (002) (I = 39.1%) and (121) (I = 70.3%), around the strongest peak (121) for the monoclinic Li3Fe2(PO4)3 phase according to the JCPDS No. 80– 1515 exist. The relative intensities of these three peaks, based on the fitting results shown in Fig. 1b, are presented in Table S1 in Supporting information. The undoped Li3Fe2(PO4)3 sample, reported by Yang et al. [18], presents the relative intensities of the three peaks almost same as the standard one. In addition to a slight left-shift of the XRD peaks due to the replacement of larger V5+ ions (0.0355 nm) for the smaller P5+ ions (0.017 nm) [32], VO43- doping greatly weakens the left (002) XRD peak. However, introduction of Ti4+ cations not only results in a right-shift of the XRD peaks caused by the lattice contraction derived from the replacement of smaller Ti4+ ions (0.0605 nm) for the larger Fe3+ ions (0.0645 nm) [32], but also sharply lessens the right (121) XRD peak, concomitant with strengthening of the left (002) peak. Those variations in XRD peaks mainly correspond to the changes in concentration of the atoms located at coordinates of (0, 0, 0.5) and (0, 0.5, 0), respectively, respect to the Li3Fe2(PO4)3 crystal. The V5+ ion is about one time larger than the P5+ ion. When the larger V5+ ions replace the smaller P5+ ones in the Li3Fe2(PO4)3 crystal, positions of the other atoms must be adjusted in addition to a slight lattice expansion. On the other hand, introduction of Ti4+ ions mainly crowds out the atoms at (0, 0.5, 0), along with re-backing to (0, 0, 0.5) for the out-crowded atoms by VO43- doping. Table S1 reveals that the number of atoms crowded out by Ti4+ doping increases with an increase in the Ti4+ contents. Those out-crowded atoms move to other positions and result in an adjustment in other parameters of the Li3Fe2(PO4)3 lattice.
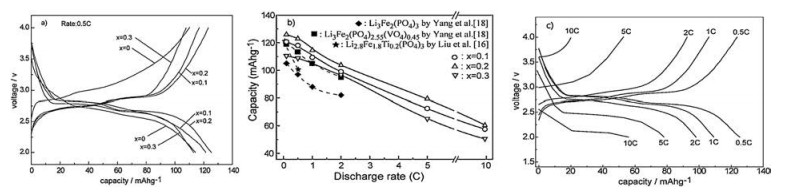
Fig. 2 shows the initial charge-discharge profiles of the samples with different Ti4+ contents, along with their rate capabilities studied in a range of cycling rates. The Ti4+-containing samples exhibit two charge-discharge plateaus longer than the only VO43-- substituted one, i.e., x = 0 or Li3Fe2(PO4)2.55(PO4)0.45. The discharging capacity increases with an increase in the Ti4+ contents, reaches the maximum at the Ti4+ content of 0.2, and then decreases with a further increase in the Ti4+ content. The initial discharging capacity is about 125.4 mAh/g at 0.5C for the sample with Ti4+ content of 0.2. This discharge value is very close to the theoretical capacity of the Li3Fe2(PO4)3 compound, and about 10.6% and 25.0% larger than those of the VO43--substituted Li3Fe2(PO4)2.55(PO4)0.45 reported by Yang et al. [18] and the Ti4+-substituted Li2.8Fe2.8Ti0.2(PO4)3 by Liu et al. [16], respectively. Though the discharging capacity decreases with an increase in discharging rates, the Ti4+-containing samples present the discharging capacity higher than the others. A high discharging capacity of 80 mAh/g is achieved at a rate of 5C for the sample with x = 0.2, this value is comparable with the value obtained at 2C rate of the Li3Fe2(PO4)3 reported by Yang et al. [18]. In comparison with the only VO43--substituted one, the sample with Ti4+ content of 0.2 also presents an apparently reduced polarization during the process of Li+ insertion and extraction (Fig. 2c). The sample with x = 0.3 exhibits lower discharge capacity than the other two samples, which should be related to the more contraction in lattice parameters caused by Ti4+ doping as shown in Fig. 1, deteriorating lithium conduction in the 3D channels. Even so, the sample of x = 0.3 presents the larger discharging capacity than the ones without Ti4+, especially at higher discharging rates (≥1C). These results suggest that simultaneous substitution of Ti4+ and VO43- greatly improves the electrochemical performance of the Li3Fe2(PO4)3 compound, and this is more effective than those doped by the single Ti4+ or VO43- substitution, especially at high discharging rates.

|
Download:
|
| Fig. 2. Initial charge-discharge profiles at 0.5C rate (a) and rate capability (b) of the samples with different Ti4+ contents, and initial charge-discharge profiles of the samples with x = 0.2 at different rates (c). | |
{kind=link}
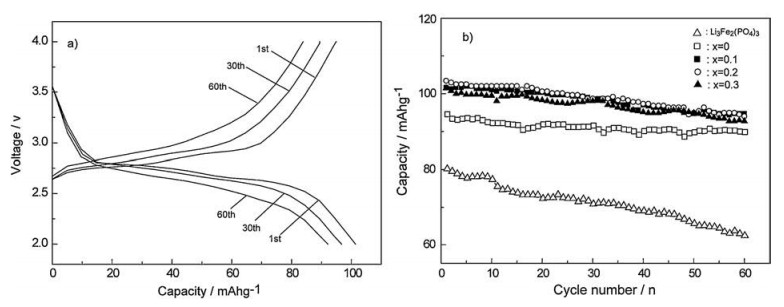
In order to further investigate the effect of the Ti4+ substitution on the Li3-xTixFe2-x(PO4)2.55(VO4)0.45 cathode material performances, the sample with x = 0.2 was galvanostatically cycled at 2C rate (Fig. 3a). One can see that the potential profiles of the cathode present two typical plateaus related to insertion of two Li+ ions. Meanwhile, the potential profiles of the material maintain similar shape and sizes upon the subsequent cycles, which confirms its good reversibility. Fig. 3b shows the cycling data of the samples with different Ti4+ contents and the bare Li3Fe2(PO4)3 cathode at 2C rate. Single VO43- substitution apparently improves the capacity retention of the Li3Fe2(PO4)3 during the charge-discharge cycles, meanwhile, introduction of the Ti4+ cations to the VO43-- substituted one further enhances its discharging capacities at the same cycling. All the Ti4+ doping samples present a similar discharging capacity at the same number of cycles. Among them, the sample with x = 0.2 could deliver the largest discharging capacity of 95.4 mAh/g after 60 cycles, indicating the superior cycling stability.

|
Download:
|
| Fig. 3. Charge-discharge profiles of the sample of x = 0.2 at 2C rate (a) and cycling performance of the samples with different Ti4+ contents (b). | |
{kind=link}
Fig. 4a shows the rate performance of the sample of x = 0.2 at room temperature, it is cycled by a mode of charging to 4 V under the same current densities as discharging to 2.0 V. Even though the discharging capacity decreases with an increase in the discharging rates from 0.5C to 10C, it is noteworthy that the cell capacity can almost recover to the initial value when the discharging rate was set back to 0.5C. Those results suggest that this sample could maintain a stable structure even at a high discharging rate. On the other hand, XRD patterns of the cathodes before and after rate cycling are shown in Fig. 4b, corresponding to points A and B in Fig. 4a, respectively. The cathode after 60 discharging-charging cycles under severe rate changing presents the almost same XRD pattern as that of the cathode before discharging (point A in Fig. 4a). This result further proves that the sample with x = 0.2 has a good stability and reversibility in our study.

|
Download:
|
| Fig. 4. Variation in discharging capacity at gradually changing rates (a), and XRD patterns (b) of the cathodes. The cathodes used the sample of x = 0.2 as the active material. | |
{kind=link}
CV curves of the samples with x = 0 and 0.2, together with the Li3Fe2(PO4)3 reported by Yang et al. [18], are shown in Fig. S1 (Supporting information). Two distinct peaks could be observed in the discharge process for all samples, which could be attributed to the insertion of two Li+ ions occurring at 2.6 V and 2.8 V regions coupled with the Fe3+/Fe2+ redox processes, the results are consistent with the observation in Figs. 2 and 3. The sample with x = 0.2 possesses higher CV peak intensities for both reduction and oxidation, and a lower electrochemical polarization than the bare Li3Fe2(PO4)3 and the single VO43--substituted one. It can be inferred that simultaneous presence of Ti4+ and VO43- in the Li3Fe2(PO4)3 compound exhibits remarkable positive effect of the electrochemical response and properties of this material as a cathode for lithium batteries.
EIS technique was employed to further investigate the effect of simultaneous substitution of Ti4+ and VO43- on electrochemical performance of the Li3Fe2(PO4)3 in a lithium half-cell with different composition cathode. The EIS data are presented in Fig. S2a (Supporting information). Nyquist plots of all studied samples display the same features and consist of a compressed semicircle in the high-to-medium frequency range and a straight line in the low frequency range. While the low frequency straight line is attributed to the "Warburg" impedance" resembling the solid state diffusion of Li ions within the solid active mass, the compressed semicircle is mainly attributed to the charge-transfer impedance at the electrode/electrolyte interface. It can be seen that the charge transfer resistance decreases with an increase in the Ti4+ content, and approaches its minimum at x = 0.2. This minimum value is only about 65% and 30% of those for the single VO43--substituted sample and the bare one, respectively. Charge transfer conditions are critical for the electrode performance, especially at the elevated densities. Based on the VO43- anion substitution, the Ti4+ cation substitution further reduces the charge transfer impedance and contributes to the cathode performance enhancement in a lithium cell, which was clearly observed from the experimental data above on battery cycling and CV curves.
The diffusion coefficient of lithium ion, D, can be calculated using the following Eq. (1) [33]:

|
(1) |
Where R is the universal gas constant, T is the absolute temperature, n is the surface area of electrode, F is the Faraday constant, C is the molar concentration of Li+, and σ is Warburg coefficient, which could be defined from the Eq. (2) [33]:

|
(2) |
where Zre is the real part of the EIS, Rs is the electrolyte resistance, Rct is the charge-transfer resistance and v is the angular frequency in the low frequency region, as shown in Fig. S2b (Supporting information). The obtained D value for all the studied cathodes are presented in Table S2 (Supporting information). One can see from those data that the sample of x = 0.2 has the largest value of D while the bare Li3Fe2(PO4)3 and the only VO43- substituted sample own the small lithium-ion diffusion coefficient. Charge-transfer diffusion is crucial for the materials applicability for practical purposes, as it determines the electrochemical processes kinetics. It can be seen that simultaneous presence of Ti4+ and VO43- used in the current work remarkably improves the Li-ion diffusion in the system. Along with the results presented above, it proves that substitution of the Ti4+ cation for Fe3+ and VO43- anion for PO43- as a powerful technique to improve the electrical conductivity of the Nasicon Li3Fe2(PO4)3 compound and its overall electrochemical performance as a cathode for lithium batteries.
Although ionic conductivity is known to be much less than electronic conductivity for the electrode materials, the ionic conductivity is crucial for progress of electrochemical reactions in the electrode. Different from other electrode materials, long-range conduction for the mobile ions in the Nasicon structure involves hopping between M1 and M2 site sites, which are joined by a bottleneck. On the other hand, researches on Nasicon solid electrolytes reveal that movement of the mobile ions along the 3D conduction channels strongly depends on their structural parameters or the bottleneck size between M1 and M2 sites [30, 34]. XRD results shown in Fig. 1 and Table S1 suggest that the Li3Fe2(PO4)3 with Nasicon structure changes its bottleneck size between M1 and M2 sites due to variation in the lattice parameters and the atom concentration at different positions resulting from the VO43- and/or Ti4+ doping, and then to affect its electrochemical properties. Furthermore, difference electronegativity between Ti (1.54) and Fe(1.83), as well as V(1.63) and P(2.19) [35], can alter the local electron density for the nearly iron and phosphorus, and then the ion conduction through them. The above electrochemical data suggest that the sample with x = 0.2 presents the most appropriate bottleneck size for lithium ion conduction.
In this work, Ti4+ cations were used to replace partial Fe3+ ions in the VO43- -substituted Li3Fe2(PO4)2.55(VO4)0.45forthe preparationof high performance cathode for lithium ion batteries. Inaddition to the main Nasicon compound, some secondary phases were formed in the products by introduction of Ti4+ ions. Electrochemical measurements proved that simultaneous presence of Ti4+ and VO43- in the Li3Fe2(PO4)3 remarkably enhanced its rate capability and cycling performance via the improved conductivity and lithium diffusion, favoring releasing the stresses generated by the repetitive Li+ intercalations/extraction, which consequently led to a lower electrochemical polarization at high rate charge-discharge process. Among the Ti4+ substituted samples, the sample with x = 0.2 exhibits the best electrochemical performance delivering a high initial capacity of 125.4 mAh/g at 0.5C rate and 102.6 mAh/g at a capacity retention of 93% after 60 cycles at 2C rate. Rate performance measurements between 0.5C to 10C proved that this sample possessed a good stability and reversibility. Those performance enhancements prove the partial substitution of iron and phosphorus by titanium and vanadium in Li3Fe2(PO4)3 as a beneficial technique to remarkably improve reversible intercalation/extraction of Li+; and the electrochemical properties exhibited bythe sample with x = 0.2 prepared in this wokemake it a promising cathode for high performance lithium-ion batteries.
AcknowledgmentThis work was supported by Natural Science Foundation of Hebei Province (No. E2016202358).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2018.11.004.
| [1] |
D. Morgan, G. Ceder, M.Y. Saidi, et al., J. Power Sources 119-121 (2002) 755-759. |
| [2] |
X. Rui, Q. Yan, M. Skyllas-Kazacos, T.M. Lim, J. Power Sources 258 (2014) 19-38. DOI:10.1016/j.jpowsour.2014.01.126 |
| [3] |
J. Shirakawa, M. Nakayama, M. Wakihara, A. Uchimoto, J. Phys. Chem. B 111 (2007) 1424-1430. DOI:10.1021/jp065802g |
| [4] |
F. Fabian, M. Sugnaux, C. Savy, et al., J. Appl. Energy. Soc. 230 (2018) 1633-1644. DOI:10.1016/j.apenergy.2018.09.030 |
| [5] |
S. Patoux, C. Wurm, M. Morcrette, G. Rousse, C. Masquelier, J. Power Sources 119 (2003) 278-284. |
| [6] |
D.L. Xiao, J. Tonga, Y. Feng, et al., Solid State Ion. 324 (2018) 202-206. DOI:10.1016/j.ssi.2018.07.011 |
| [7] |
D.L. Smiley, L.J.M. Davis, G.R. Goward, J. Phys. Chem. C 117 (2013) 24181-24188. DOI:10.1021/jp407510h |
| [8] |
S. Zhu, H. Zhou, T. Miyoshi, et al., Adv. Mater. 16 (2004) 2012-2017. |
| [9] |
R. Younesi, A. Christiansen, S. Loftager, et al., ChemSusChem 8 (2015) 3213-3216. DOI:10.1002/cssc.201500752 |
| [10] |
L.J.M. Davis, I. Heinmaa, G.R. Goward, Chem. Mater. 22 (2010) 769-775. DOI:10.1021/cm901402u |
| [11] |
M. Morcrette, C. Wurm, C. Masquelier, Solid State Sci. 4 (2002) 239-246. DOI:10.1016/S1293-2558(01)01235-3 |
| [12] |
A.R. Shaikhlislamova, I.A. Stenina, A.B. Yeroslavtesv, Russ. J. Inorg. Chem. 53 (2008) 1829-1833. DOI:10.1134/S0036023608120012 |
| [13] |
N. Plylahan, C. Vidal-Abarca, P. Lavela, J.L. Tirado, Electrochim. Acta 62 (2012) 124-131. DOI:10.1016/j.electacta.2011.11.117 |
| [14] |
C.H. Song, J.K. Sun, F.Q. Huang, Z.Q. Liu, H.E. Pin-Gang, Funct. Mater. 4 (2009) 604-607. |
| [15] |
J.K. Sun, F.Q. Huang, Y.M. Wang, et al., J. Alloys. Compd. 469 (2009) 327-331. DOI:10.1016/j.jallcom.2008.01.109 |
| [16] |
Z. Liu, F. Huang, J. Sun, Mater. Sci. Eng. B 176 (2011) 1313-1316. DOI:10.1016/j.mseb.2011.07.009 |
| [17] |
A.K. Padhi, V. Manivannan, J.B. Goodenough, J. Electrochem. Soc. 29 (1998) 514-520. |
| [18] |
Y.G. Yang, Y.G. Zhang, Z.S. Hua, X. Wang, H.F. Peng, Electrochim. Acta 219 (2016) 547-552. DOI:10.1016/j.electacta.2016.10.044 |
| [19] |
B. Zhang, J. He, Z.S. Hua, et al., Chin. J. Inorg. Chem. 32 (2016) 19-26. |
| [20] |
B. Zhang, J. He, Z.S. Hua, X. Wang, H.F. Peng, Chem. J. Chin. U 38 (2017) 108-114. |
| [21] |
M. Tatsumisago, K. Yoneda, T. Minami, J. Am. Ceram. Soc. 71 (2005) 766-769. |
| [22] |
M. Tatsumisago, N. Machida, T. Minami, J. Ceram. Soc. Jpn. 95 (1987) 197-201. |
| [23] |
A. Magichris, G. Chiodelli, M. Duclot, Solid State Ion. 9-10 (1983) 611-615. DOI:10.1016/0167-2738(83)90303-X |
| [24] |
B. Carette, M. Ribes, J.L. Souquet, Solid State Ion. 9-10 (1983) 735-737. DOI:10.1016/0167-2738(83)90323-5 |
| [25] |
S.X. Geng, Y.G. Yang, Y.G. Zhang, et al., Electrochim. Acta 176 (2015) 327-333. DOI:10.1016/j.electacta.2015.06.047 |
| [26] |
H.F. Peng, M.L. Gao, M.L. Wang, C.X. Chen, Chin. J. Inorg. Chem. 27 (2011) 1969-1974. |
| [27] |
T. Takahashi, O. Yamamoto, J. Electrochem. Soc. 117 (1970) 1-5. DOI:10.1149/1.2407428 |
| [28] |
T. Takahashi, O. Yamamoto, S. Yamada, S. Hayashi, J. Electrochem. Soc. 126 (1979) 1335-1337. DOI:10.1149/1.2129272 |
| [29] |
A.S. Best, P.J. Newman, D.R. MacFarlane, K.M. Nairn, S. Wong, Solid State Ion. 126 (1999) 191-196. DOI:10.1016/S0167-2738(99)00212-X |
| [30] |
S. Wong, P.J. Newman, A.S. Best, et al., J. Mater. Chem. 8 (1998) 2199-2203. DOI:10.1039/a802752h |
| [31] |
X. Song, M. Jia, R. Chen, J. Mater. Proces. Technol. 120 (2002) 21-25. DOI:10.1016/S0924-0136(01)01044-5 |
| [32] |
R.D. Shannon, Acta Cryst. 32 (2015) 751-767. |
| [33] |
H.P. Liu, S.F. Bi, G.G. Wen, et al., J. Alloys. Compd. 43 (2012) 99-104. |
| [34] |
A. Martinez-Juárez, C. Pechammán, J.E. Iglesias, J.M. Rojo, J. Phys. Chem. B 102 (1998) 372-375. DOI:10.1021/jp973296c |
| [35] |
L. Pauling, The Nature of the Chemical Bond. 3rd ed. Ithaca, NY: Cornell University Press, 1960.
|