 2019, Vol. 30
2019, Vol. 30 


The production and application of dimethyl carbonate (DMC) have received extensive attention, due to its environmental friendliness as well as versatile chemical properties [1]. It can be used to manufacture polycarbonates and substitute toxic dimethyl sulfate and phosgene as methylation carbonylation reagent. Furthermore, it is also considered as a fuel additive to improve the combustion performance and reduce emissions [2, 3]. Among the various synthesis routes, the methanol oxidative carbonylation process that uses methanol, CO, and O2 as raw materials is promising alternative, due to its high atom economy (83%), favorable thermodynamics, and moderate conditions [4-6].
King first demonstrated that chlorine is not necessary in this reaction [7]. Afterwards, Cu-exchanged zeolite as a kind of chlorine-free catalyst was intensively investigated recently [8, 9]. H-form faujasite (FAU, e.g., X and Y) possesses a high content of tetrahedral Al atoms, providing a large number of Brønsted acid sites for ion exchange with Cu cations [10]. Besides, the unique framework structure provides a stable and appropriate local environment for Cu+ cations that is prone to selectively catalyze the formation of DMC [11]. Moreover, the supercages in FAU with large window and cavity (diameter of 0.74 nm and 1.12 nm, respectively) make the reactant molecules more accessible to the active Cu sites [12]. Until now, several studies have been reported to shed light on the role of the position and local environment of Cu species in oxycarbonylation of methanol. Lamberti et al. employed complementary characterizations (e.g., XRD, XAS, IR) to study the localization of Cu+ in CuY that is prepared by solid state ion exchange (SSIE) using NH4-Y and CuCl. The results show that 23.4 Cu+/unit cell (u.c.) occupy site Ⅰ, and 6.1 Cu+/u.c. and 11.5 Cu+/u.c. are located at site Ⅱ and Ⅱ* (Fig. S1 in Supporting information) [13]. Bell et al. combined the XAS and IR characterization to study CuY catalyst and found that only the Cu+ species at site Ⅱ and Ⅲ are accessible to the reactant molecules [14]. Our previous work suggested that the Brønsted acidity of zeolites, which presents in large cavities and accessible to pyridine molecule dominates the catalytic performance of Cu-exchanged zeolites on oxidative carbonylation quantitatively [15]. Actually, 60% of Al atoms are located at sodalite cages with 6-membered ring window of 0.25 nm [11]. This indicates that a large proportion of Cu species are not catalytically active in this reaction, taking into diffusion restriction into consideration. Therefore, improving the accessibility of Cu species by modifying the porosity of FAU zeolite is a strategy to enhance the catalytic properties of Cu-FAU zeolites.
Post-treatment has been extensively used to varying the microporosity of zeolite. However, alkaline treatment usually does not work because of repulsion between OH- and the negatively charged framework [16], while acidic and steam treatment are prone to dealumination that decrease Brønsted acidity [17]. Herein, we employed a method to etch zeolite using NH4F by means of its double hydrolysis to HF2-, according to the reference [18]. The influence of etching on porosity was investigated by combining several adsorption experiments of different probe molecules (e.g., NH3, CO, pyridine). As a result, the activity of Cu-Y was enhanced, owing to the changes in accessibility of Cu species.
A NH4Y zeolite that was obtained from ion-exchange of commercial NaY (SiO2/Al2O3=5) with NH4Cl in solution, was used as the parent catalyst support. The etching treatment was carried out in liquid (the mass ratio of 25wt% NH4F solution to NaY=6) with ultrasonic assistant for 0, 5, 10 and 20min in an ice bath. The slurry was washed with hot deionized water, followed by drying at 80 ℃ under vacuum overnight and calcination at 550 ℃ for 3h in muffle oven. The obtained samples were labeled as HY-x, in which x represents the etching time. CuY catalysts were prepared by solid-state ion exchange of HY with purified CuCl (HY:CuCl=1:0.3, g/g) at 550 ℃ in a flowof N2 for 8h. The final samples were denoted as CuY-x.
N2 adsorption-desorption was used to explore pore structure of HY catalysts. All the samples show characteristic of microporosity (Fig. 1A). Table 1 summarized the specific surface areas, micropore and mesopore volumes that were calculated by the BET and t-plot method respectively. Compared with the reference HY-0, the surface area as well as micro- and meso- porosity was increased with extending the etching time. Pore distribution shows that the pore size mainly centers at 8Å and 12Å (Fig. 1B), approximately consistent with the size of super- and sodalite- cages respectively. With the increase of etching time, the micropores with a pore size of 8Å obviously increases and then decreases. As the diameter of sodalite cages is 0.63nm, we deduce that the sodalite cages might be opened by this post-treatment. The pore volume reaches the highest value when the HY was etched in NH4F solution for 10min. Further prolong the etching process to 20min, the specific surface area and micropore volume decreases, implying a significant collapse of zeolitic framework, which is consistent of crystallinity obtained from XRD patterns. ICP results show that the ratio of Si/Al is almost maintained (Table 1), because the in situ generated HF2- species in NH4F solution extracts framework Si and Al cations at equal rates [18].

|
Download:
|
| Fig. 1. (A) Nitrogen adsorption–desorption isotherms and (B) pore distribution of the HY-x catalysts (C) NH3-TPD profiles of HY-x samples; (D) FTIR spectra of pyridine adsorbed on HY-x samples; (E) FTIR spectra of CO adsorption; (F) Relationship between Brønsted acid content and Cu(I) content. | |
|
|
Table 1 Physical and chemical properties of HY-x samples. |

In order to explore the effect of the etching process on the structure of the catalysts, XRD was performed and the results are shown in Fig. S2 in Supporting information. All the samples show characteristic diffraction peaks of pure zeolite Y phase. Note that the intensity of the peaks becomes weaker as the etching time increases. The calculated relative crystallinity was given in Table 1 by comparing the sum areas of all the characteristic peaks of modified samples with that of the parent HY-0 sample [19]. It is obvious that the etching process results in a loss of crystallinity, which becomes severer with longer time.
Transmission Electron Microscope (TEM) image of the CuY-x catalysts (x=5, 10, 20) shows that there are amorphous species around the zeolite particle, demonstrating solution of zeolite framework (Fig. S3 in Supporting information). This is in agreement with the partial loss of crystallinity obtained from XRD. Furthermore, some defects and interruptions of the lattice planes are visible, suggesting local extraction of T atoms in framework. This observation implies the possibility of the opening of cages in zeolite Y. We also found that no visible Cu species exist, indicative of high dispersion of Cu species. So we can conclude that most of CuCl exchanged with the Brønsted acidic protons and the formed isolated Cu+ cations are bonded to the framework O atom.
As Brønsted acid in zeolite provides exchangeable sites for Cu+ during SSIE to prepare CuY catalysts, we use basic probe molecules of different size to quantitatively determine the acidity, including number and accessibility. NH3-TPD was performed to determine the total number of acid sites on HY-x samples (Fig. 1C). The similarity of the curves for different samples indicates little effect of the etching process on acidity. As listed in Table 1, it is obvious that the total numbers of acid sites on the samples are almost the same. These observations coincide with the Si/Al ratio determined by ICP-OES. Pyridine, as a probe molecule, is sensitive in FTIR to distinguish the Brønsted and Lewis acid sites. Besides, the diameter of pyridine molecule is about 5 Å, much bigger than the size of the six-membered ring (2.2~2.6 Å). Therefore, taking the molecular size into account, only acid sites located in supercages (e.g., at site Ⅱ and Ⅲ) can interact with pyridine because of steric hindrance, while the acid sites in sodalites and prisms units are supposed to be inaccessible to pyridine [15]. The FTIR spectra of pyridine adsorption of the four samples are shown in Fig. 1D. The bands near 1450 cm-1 and 1610 cm-1 are attributed to the pyridine interacted with Lewis acid sites, the 1540 cm-1 and 1630 cm-1 bands are associated with pyridinium ion on Brønsted sites, and the 1490 cm-1 absorption peak is due to the contribution of both Lewis and Brønsted acid sites [9]. We calculated the number of Brønsted acid sites based on the integral area of the band at 1540 cm-1, according to the ref. [20] The results listed in Table 1 demonstrates that the number of Brønsted acid sites detected by pyridine is increased first and then decreased with increasing the etching time. It implies that the pyridine molecules are allowed to enter the cages with smaller size such as sodalite cages and interact with more Brønsted acid sites. Combining with the N2 adsorption isotherms, it is clear that NH4F etching opens the sodalite cages, improving the accessibility of Brønsted acid sites. As the size of pyridine molecule is larger than that of the reactants (i.e., CO, methanol and O2), it is reasonable to infer that the opening of sodalite cages will allow the diffusion of the reactant molecules into sodalite cages and reaction on more Cu+ sites.
CO is widely used as a probe to determine the nature of metal species in catalysis, not only the quantity but also the location and oxidation state. To ensure the influence of the opening of cages on accessibility of Cu+ active sites, we performed irreversible CO adsorption as well as CO adsorption IR. According to the literature, CO would be prevented to go into the sodalite cages due to its molecular size [12, 21]. The accessible Cu+ sites were calculated from the irreversible CO adsorption by assuming that one molecule of CO was adsorbed on one Cu+ cation. As illustrated in Table 2, we can easily find that the amount of available Cu+ sites for CO adsorption varied as the same trends as N2 adsorption isotherms and pyridine adsorption IR. This result strongly confirms the improved accessibility of Cu+ active sites, stemming from the opening of the small cages during NH4F etching. In other words, it is expected that the amount of available Cu+ sites at the same Cu loading is increased. We also noted that the calculated available Cu+ sites are lower than actually loading of Cu obtained by ICP-OES. This indicates that there are still a part of Cu+ located at sodalite and hexagonal positions even after etching treatment, which is catalytically inactive in this reaction. CuY-10 has the highest amount of Cu(I). Further increasing the etching time, the Cu(I) content has a light decrease, might because of a collapse of framework. In order to further certify the conclusion of irreversible CO adsorption, CO adsorbed FTIR experiments are conducted. The normalized spectra of CO adsorption are illustrated in Fig. 1E. The overlapped two bands at 2160 cm-1 and 2146 cm-1 are attributed to CO adsorbed on Cu+ cations at site Ⅱ* and Ⅱ, respectively [13]. The variation tend of the band intensity is solid to support the results of irreversible CO adsorption. We plotted the calculated Cu+ content versus the number of Brønsted acid sites detected by pyridine (Fig. 1F), and the result show a linear relationship between them. The CuCl exchanges with the Brønsted acid sites to form Cu+ sites during the SSIE. As the CO is much smaller than pyridine, it is reasonable that if the Brønsted acid sites are detectable to pyridine, the Cu+ are accessible to CO.
|
|
Table 2 Nature and local environment of Cu species in different characterizations. |
{kind=link}
Based on these characterizations, it can be concluded that partial of small cages are opened through NH4F etching, resulting in an improvement of Cu+ sites accessibility.
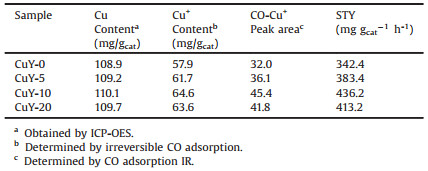
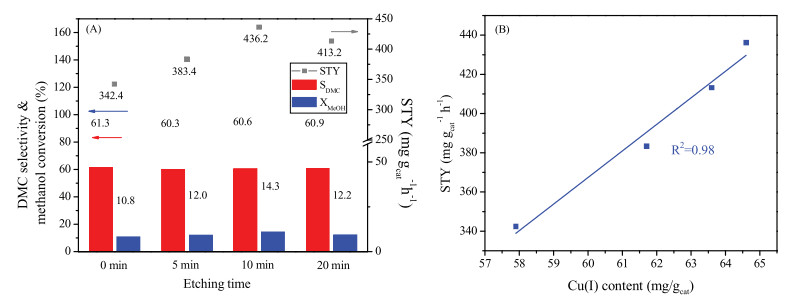
The catalytic performances of the CuY catalysts on oxidative carbonylation of methanol were evaluated in a continuous fix-bed microreactor system and the products were analyzed by online gas chromatography. Fig. 2A shows the space-time yield (STY) and selectivity of DMC as well as methanol conversion over CuY samples treated with different etching time. It is found that all the CuY-x (x = 5, 10, 20) catalysts using the modified HY zeolite as supports exhibit higher activities in comparison with CuY-0, stemming from more accessible Cu+ sites. The CuY-10 shows the best activity with STY and conversion of 436.2 mg gcat-1 h-1. Meanwhile, the selectivity to DMC keeps a nearly constant value of ~60%. As the Brønsted acid sites of zeolite usually function as the active sites for the side-reaction, such as the decomposition of DMC [22], the similar selectivity also indicates the same exchange level of Cu+ sites. Then, we plotted the STY of DMC versus the content of Cu+ species based on irreversible CO adsoprtion in Fig. 2B It is obvious that there is a positive linear relationship between them. This result indicated that the opening of the partial cages enabled the reactant molecules such as CO, methanol to diffuse into the smaller cages of zeolite Y. And more exchanged Cu+ sites become accessible and catalytically active. In addition, the catalytic activities on CuY-x with or without etching remained substantially stable within 12 h during the reaction (Figs. S4 and S5 in Supporting information).

|
Download:
|
| Fig. 2. (A) Catalytic performances of different CuY catalysts prepared with different etching time. (Reaction conditions: 0.7 MPa, 140 ℃, total flow rate = 81.2 mL/min, time on stream = 2 h, methanol:CO:O2:N2 = 4.8:12.8:1:9.4); (B) Relationship between Cu(I) content and catalyst activity. | |
{kind=link}
In summary, the post-treatment by NH4F etching was used to modify the pore structure of zeolite Y. Physical and chemical adsorption of several probe molecules with different sizes show that the small cages are opened with keeping the zeolitic framework, resulting in a decreased diffusion limitation of reactant molecules. Therefore, the originally wasted "active center", Cu+ cations in small cages were utilized, leading to an enhanced activity. The influence of etching time was also investigated. When the etching time reached 10 min, the improved accessibility of Cu+ sites as well as maintained structure is responsible for the highest yield of DMC (436.2 mg gcat-1 h-1).
AcknowledgmentThe financial supports from the National Natural Science Foundation of China NSFC, Nos. U1510203, 21406120, 21325626 are gratefully acknowledged.
Appendix A. Supplementary dataSupplementary material related to this article can be found, inthe online version, at doi: https://doi.org/10.1016/j.cclet.2018.10.005.
| [1] |
N. Keller, G. Rebmann, V. Keller, J. Mol. Catal. A:Chem. 317 (2010) 1-18. DOI:10.1016/j.molcata.2009.10.027 |
| [2] |
S.Y. Huang, J.J. Zhang, Y. Wang, et al., Ind. Eng. Chem. Res. 53 (2014) 5838-5845. DOI:10.1021/ie500288g |
| [3] |
T.J. Fu, X. Wang, H.Y. Zheng, Z. Li, Carbon 115 (2017) 363-374. DOI:10.1016/j.carbon.2017.01.004 |
| [4] |
S.Y. Huang, B. Yan, S.P. Wang, X.B. Ma, Chem. Soc. Rev. 44 (2015) 3079-3116. DOI:10.1039/C4CS00374H |
| [5] |
S.A. Anderson, T.W. Root, J. Catal. 217 (2003) 396-405. DOI:10.1016/S0021-9517(02)00159-8 |
| [6] |
S.T. Hong, H.S. Park, J.S. Lim, et al., Res. Chem. Intermed. 32 (2006) 737-747. DOI:10.1163/156856706778606552 |
| [7] |
S.T. King, Catal. Today 33 (1997) 173-182. DOI:10.1016/S0920-5861(96)00118-6 |
| [8] |
Z. Li, K.C. Xie, R.C.T. Slade, Appl. Catal. A 209 (2001) 107-115. DOI:10.1016/S0926-860X(00)00745-6 |
| [9] |
H.M. Zhan, S.Y. Huang, Y. Li, et al., Catal. Sci. Technol. 5 (2015) 4378-4389. DOI:10.1039/C5CY00460H |
| [10] |
Y.H. Zhang, A.T. Bell, J. Catal. 255 (2008) 153-161. DOI:10.1016/j.jcat.2008.01.033 |
| [11] |
I.J. Drake, Y.H. Zhang, D. Briggs, et al., J. Phys. Chem. B 110 (2006) 11654. |
| [12] |
S. Bordiga, E. Garrone, C. Lamberti, et al., J. Chem. Soc. Faraday Trans. 90 (1994) 3367-3372. DOI:10.1039/FT9949003367 |
| [13] |
G.T. Palomino, S. Bordiga, A. Zecchina, G.L. Marra, C. Lamberti, J. Phys. Chem. B 104 (2000) 8641-8651. DOI:10.1021/jp000584r |
| [14] |
Y.H. Zhang, D.N. Briggs, E.D. Smit, A.T. Bell, J. Catal. 251 (2007) 443-452. DOI:10.1016/j.jcat.2007.07.018 |
| [15] |
S.Y. Huang, Y. Wang, Z.Z. Wang, et al., Appl. Catal. A s417- 418 (2012) 236-242. |
| [16] |
D. Verboekend, J. Pérez-Ramírez, Catal. Sci. Technol. 1 (2011) 879-890. DOI:10.1039/c1cy00150g |
| [17] |
P. Matias, J.M. Lopes, P. Ayrault, et al., Appl. Catal. A 365 (2009) 207-213. DOI:10.1016/j.apcata.2009.06.014 |
| [18] |
Z.X. Qin, K.A. Cychosz, G. Melinte, et al., J. Am. Chem. Soc. 139 (2017) 17273-17276. DOI:10.1021/jacs.7b10316 |
| [19] |
S.Y. Huang, P.Z. Chen, B. Yan, et al., Ind. Eng. Chem. Res. 52 (2013) 6349-6356. DOI:10.1021/ie3032235 |
| [20] |
C.A. Emeis, J. Catal. 141 (1993) 347-354. DOI:10.1006/jcat.1993.1145 |
| [21] |
S. Bordiga, D. Scarano, G. Spoto, et al., Vib. Spectro. 5 (1993) 69-74. DOI:10.1016/0924-2031(93)87056-Y |
| [22] |
S.A. Anderson, S. Manthata, T.W. Root, Appl. Catal. A 280 (2005) 117-124. DOI:10.1016/j.apcata.2004.10.003 |