 2019, Vol. 30
2019, Vol. 30 


b Key Laboratory of Organo-pharmaceutical, Chemistry Institution, Gannan Normal University, Ganzhou 341000, China;
c College of Chemical Engineering, Ningbo University of Technology, Ningbo 315211, China
Tetrathiafulvalene (TTF) and its derivatives have paid much attention due to the strong electron-donating ability which could be used as building blocks to construct new functional materials [1, 2]. The unique planar conjugated structure, good electronic property and reversible redox property make such organic materials widely used in photoelectric switches, molecular devices, and sensors [3-6]. By modifying the TTF core, TTF derivatives with different functional groups and structures can be obtained. In addition to the S…S interaction between sulfur atoms of TTF core in short range, hydrogen bonding and π-π interactions also have the chance to interact with each other, so that these compounds can self-assemble to form ordered structures.
Among them, the TTF derivatives with pyridine substituent groups have been widely studied [7-9]. The pyridine group can be directly attached to the TTF core without any spacer part to increase the electronic coupling between the TTF moiety and the pyridine group. The pyridine group can also well combine with the proton to form hydrogen bonding, which provides a favorable condition for the construction of supramolecular structure. Besides, the N atoms on pyridine group are potential coordination units which can be widely used in nonlinear optics, probes and molecular sensing materials [10-12]. The TTF derivative containing pyridine group as an electron donor can be used to study the intramolecular charge transfer. And they can form a complex with the transition metal to form potential electromagnetic materials [13, 14]. Therefore, it is of great importance to study their arrangement, morphology, molecular orientation and local structural defects on the surface of conductive substrate. These factors may affect the rate of charge transfer and even change the transfer mode of the carrier [15, 16]. The self-assembly of TTF derivatives at the liquid/solid interface has been studied through scanning tunneling microscope (STM) characterization. For example, the self-assembled structures of a series of TTF derivatives with different substituent groups, numbers of alkyl chains and lengths of alkyl chain have also been studied [17]. It can be seen that the TTF derivatives with long alkyl chain easily form long linear and regular self-assembly structure due to the strong van der Waals interaction between alkyl chains and between long alkyl chains and highly oriented pyrolytic graphite (HOPG) substrates. The solvent could affect the lattice parameters of self-assemblies of TTF derivatives with longer alkyl chain, while has little effect on the self-assemblies of TTF derivatives with shorter alkyl chain [17]. The self-assembled image of TTF derivative molecules without long alkyl chains cannot be clearly observed by STM [18]. The behavior of the TTF core in the selfassembly structure is still worth further exploring.
We previously found that 4-pyridyl-(ethylenedithio)TTF (EDTTF) could co-assemble into a brand hexagonal network with 1, 3, 5-tris(10-carboxydecyloxy)-benzene (TCDB) at the 1-phenyloctane (PO)/HOPG interface under ambient condition [19]. But, the self-assembly driving force is very weak in that coassembly system, as a result, the nanoporous network would transform into a more stable line structure. In order to obtained a stable grid structure using the small TTF derivative, two TTF derivatives para-TTF and meta-TTF both incorporating two pyridine groups but different positions of N in pyridine were designed, as shown in Scheme 1. In contrast to the EDTTF molecule with only one pyridine group attached to the TTF core, two pyridine groups in para-TTF and meta-TTF would provide a favorable method to construct supramolecular structure because of the enhanced self-assembly driving force. The results showed that para-TTF itself could assemble into an unique network at the PO/HOPG interface, while meta-TTF could not form a network. At the heptanoic acid (HA)/HOPG interface, the para-TTF and metaTTF molecules organized into regular linear assembly. In addition, the para-TTF adsorbed atop the alkyl chains of TCDB, and meta-TTF co-assembled into an independent hexagonal network, which could be holding for long time.

|
Download:
|
| Scheme 1. Chemical structures of TTF-based molecules para-TTF and meta-TTF. | |
{kind=link}
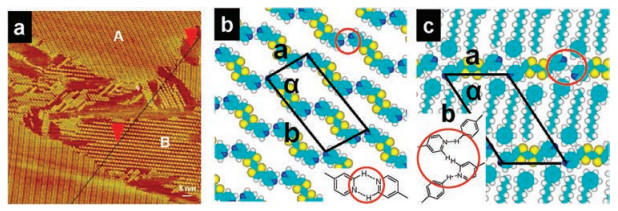
The molecular structures of TCDB and solvent such as PO and HA are shown in Fig. S1 (Supporting information). The materials used in this work, the preparation and investigation method are presented in supporting information. Firstly, the self-assembly of para-TTF or meta-TTF on HOPG surface have been studied. The large-scale para-TTF nanostructure is shown in Fig. 1a. As shown in Fig. S2a (Supporting information), the cross-sectional profile corresponding to the black solid line in Fig. 1a indicates that para-TTF lays on the surface to form monolayer. The length of each bright point is approximately 1.52 nm, which is in accordance with the size of one para-TTF molecule. In combinationwith the size and electron density information of the bright spots, it can be deduced that each bright spot should correspond to one para-TTF molecule. Two kinds of patterns denoted as domain A and domain B coexist. In the closely packed domains A, para-TTF arranged very close with compacted linear pattern. In domains B, there is a clear gap between two para-TTF aligned rows is filled with PO molecules. And high-resolution self-assembly structure of domains A and B is shown in Fig. S2b and c (Supporting information), respectively. The distance between two adjacent para-TTF molecules is estimated to be 0.2 nm, which is in accord with the N…H distance proving that the contacts can indeed be hydrogen bond. Therefore, para-TTF connects with adjacent para-TTF via a pair of C=N…H—C hydrogen bonding through two pyridine groups marked with red circle in domains A. In domain B, the hydrogen bonding between neighboring para-TTF molecules is absent. The benzene ring of solvent PO incorporates interaction with the pyridine of para-TTF molecule and forms one hydrogen bonding as circled region in Fig. 1c. Furthermore, van der Waals interactions between the adjacent PO molecules should enhance the stability of the molecular packing in domain B.

|
Download:
|
| Fig. 1. (a) Self-assembly of para-TTF molecules at PO/HOPG interface. Tunneling condition: Iset = 299.1 pA, Vbias = 599.1 mV. (b) and (c) represent calculated molecular model of pattern A and pattern B, respectively. Hydrogen bondings were marked out by red circles. Unit cells were superimposed onto STM images and molecular models of pattern A (a = 1.4 ± 0.1 nm, b = 2.8 ± 0.1 nm, α = 79 ± 2°) and pattern B (a = 1.8 ± 0.1 nm, b = 2.7 ± 0.1 nm, α = 57 ± 2°). The parameters were listed in Table S1 (Supporting information). | |
{kind=link}
The molecule para-TTF/PO formed two kinds of structures in domain A and domain B, in which PO is absent and present in the coassembly with para-TTF, respectively. The total energy per unit area of domain A and domain B is -0.235 kcal mol-1 Å-2 and -0.348 kcal mol-1 Å-2, respectively, and indicating that the domain B is the most energetically favorable pattern. Unexpectedly, meta-TTF itself could not assemble into a network at the PO/HOPG interface. Compared with para-TTF molecule, the change of N atom position in pyridine of meta-TTF molecule causes th echange of steric hindrance between molecules. And these weak molecular interactions are quite small and their assembly structure cannot exist stably.Itmight be easier to move meta-TTF molecules because N atoms are on both sides of the meta-TTF molecule, so the assembly of meta-TTF molecules is not as simple as we think.
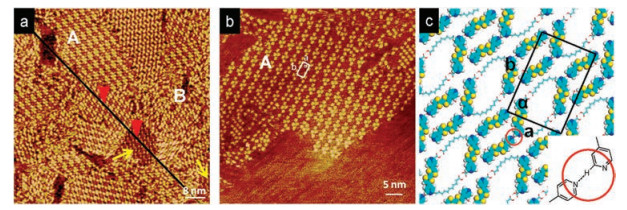
These two molecules were used to adjust the TCDB H—bonding networks. The TCDB molecule was firstly characterized by STM at the PO/HOPG interface and the traditional TCDB molecular template with rectangular nano cavity was obtained. Next, the PO solution containing the para-TTF molecule is added to the HOPG surface bearing the TCDB meshes. Fig. 2 shows a large scale STM image of the para-TTF/TCDB system with many domains of selfassembled structures exsiting. The region pointed by the yellow arrow is the traditional assembly structure of TCDB. The region A corresponds to the para-TTF/TCDB co-assembly structure controlled by the TCDB template. The region B corresponds to the confusion structures. It should be noted that the regular selfassembly structure of para-TTF itself is brokenwith the coexistence of para-TTF and TCDB molecules. Fig. 2b is a high resolution STM image corresponding to the region A. The length of TTF molecule is exactly the same as that of the alkyl chains of TCDB molecule. Fig. S3a (Supporting information) is a profile of the structure corresponding to a straight line in Fig. 2a, indicating that the structure of the para-TTF/TCDB co-assembly is obviously higher than that of the TCDB self-assembly structure. The alkyl chain of TCDB can provide TTF site for adsorption, and the carboxyl group at the end of alkyl chain also has the chance to form hydrogen bonding with guest molecules.

|
Download:
|
| Fig. 2. (a) Large-scale STM image of para-TTF/TCDB system. The yellow arrow marked out the traditional TCDB pattern. Tunneling condition: Iset = 299.1 pA, Vbias = 599.1 mV. (b) A high-resolution STM image of pattern A. Tunneling condition: Iset = 349.1 pA, Vbias = 599.1 mV. (c) Calculated molecular model of para-TTF/TCDB system. A unit cell (a = 2.5 ± 0.1 nm, b = 3.9 ± 0.1 nm, α = 94 ± 2°) was superimposed onto para-TTF/TCDB pattern with the corresponding parameters shown in Table S3 (Supporting information). Hydrogen bonding C—H…N=C was marked out by the red circle. | |
{kind=link}
Fig. 2c is the corresponding molecular model of the region A. Two para-TTF molecules are connected through weak —C—H…N=C H—bonding interaction as marked in the red circle. The para-TTF molecules are adsorbed on the fully expanded alkyl chains of TCDB. The energy per unit area of the system is calculated to be -0.332 kcal mol-1 Å-2, while that of the traditional TCDB self-assembly structure is about -0.286 kcal mol-1 Å-2 [19]. In comparison with the energy per unit area of domain A and domain B, it shows that under the regulation of TCDB template, the regular self-assembly structure para-TTF has been broken. Some para-TTF molecules will form new structure with TCDB, while the other para-TTF molecules tend to maintain the original self-assembly structure. These two co-existing structures constitute the chaotic structure shown in the region B. The results of theoretical calculation coincide with the experimental phenomena.
Fig. 3 shows a large scale STM image of the meta-TTF/TCDB system. The result is similar to the experimental results of EDTTF molecules [19]. Two kinds of patterns denoted as domain Ⅰ and domain Ⅱ can be observed. Domain Ⅰ shows the traditional pure assembly structure of TCDB. While in domain Ⅱ, a new hexagonal network is observed. Some defects with missing bright point shown by the yellow arrow in Fig. 3b could be visualized in the large STM image. As expected, meta-TTF molecules are involved in the process of large scale self-assembly. In domain Ⅱ, each hexagonal ring consists of six bright points. Since the length of the bright point is about 1.5 nm, we suggest six meta-TTF molecules form a hexagonal ring. It is interesting that all the hexagonal rings are independent of each other. The edge length of the hexagonal network is about 2.1 nm, and the size of nanopores is obviously reduced compared with those formed by EDTTF. And, the hexagonal network still existed even after the sample was holding for 48 h. We suggested that in the meta-TTF/TCDB system the three alkyl chains of TCDB were fully extended.

|
Download:
|
| Fig. 3. (a) Coexistence of traditional TCDB self-assembly (Ⅰ) and meta-TTF/TCDB system (Ⅱ). Tunneling condition: Iset = 299.1 pA, Vbias = 599.1 mV. (b) A high-resolution STM image of meta-TTF/TCDB pattern. Tunneling condition: Iset = 349.1 pA, Vbias = 599.1 mV. The distance of L = 2.1 ±0.1 nm. The yellow arrows point out the missing bright points in the packing pattern. (c) Calculated molecular model of meta-TTF/TCDB system. A unit cell (a = 4.4 ± 0.1 nm, b = 4.4 ± 0.1 nm, α = 60 ± 2°) is superimposed onto meta-TTF/TCDB pattern with the corresponding parameters shown in Table S3. | |
{kind=link}
As a result, the meta-TTF molecules can be fixed in the TCDB network through O-H…N hydrogen bond between the carboxyl group of TCDB and pyridine from meta-TTF, as placed in Fig. 3c. The total energy per unit area of meta-TTF/TCDB is -0.338 kcal mol-1 Å-2. The theoretical results also support the stable existence of nanoscale ring structure, which coincides with the experimental phenomena that the nanopores can be observed even the sample was shelving for a long time. From the co-assembly structure, we could conclude that meta-TTF molecules are not embedded in the nanopores of TCDB templates, but it interacts with TCDB to form a new hexagonal network. A cross-section profile corresponding to Fig. S3b (Supporting information) shows that the height of the bright spots is higher than that of traditional TCDB network. Therefore, we could conclude that the bright spots correspond to meta-TTF molecules due to the height and as well as the strong electronic density of the core TTF of meta-TTF.
Actually, the TCDB network can adjust itself in response to different molecular size and shape of the guest [20-22]. In addition, great changes for the size and the shape of TCDB cavity have happened during the coordination process [23]. Unlike the reported works, the size and shape of these two TTF are the same, and only the position of pyridine is different. So, it would be an interesting work to further investigate why para-TTF preferentially absorbed on the top of the alkyl chains of TCDB while meta-TTF preferentially interact with terminal carboxylic acid group of TCDB. Many small molecules with unique properties will not be able to form stable assembly in a wide range because of intermolecular interaction and weak interaction between molecules and substrates. It is also possible that in the electric field, there is an exclusion between the molecules and the STM probes, therefore the self-assembly structure of the molecule cannot be characterized by STM.
In these two systems para-TTF/TCDB and meta-TTF/TCDB, we presume PO molecules are not involved in self-assembly. Therefore, solvent do not play a major role in the self-assembly process. To further verify this idea, we dissolved TCDB and TTF molecules into the other high boiling point solvent DMF, which excludes the effects of benzene ring and alkyl chains. The STM experiments were carried out after the DMF was naturally dried in order to exclude the effect of solvent. As a result, it is found that the similar para-TTF/TCDB and meta-TTF/TCDB structures still exist, proving that solvent molecules are not involved in the selfassembly structure. The self-assembled meta-TTF/TCDB structures of at the gas/solid interface is shown in Fig. S4 (Supporting information).
In order to further investigate the effect of pyridine position on the self-assembly, one carboxylic acid solvent HA was used as solvent. And high-resolution self-assembly structure of para-TTF/ TCDB and meta-TTF/TCDB is shown in Figs. S5a and c (Suppporting information), respectively. The para-TTF and meta-TTF molecules have regular linear assembly on the HOPG interface with HA. It is concluded from the intermolecular distance that the HA molecules have been involved in the self-assembly process. However, the selfassembly structure of these two molecule is different because of the different positions of N atoms in the pyridine ring. The lattice parameters for para-TTF/HA system and meta-TTF/HA system are measured to be a = 1.9 ± 0.1 nm, b = 1.9 ± 0.1 nm, α = 59 ± 2° and a = 1.8 ± 0.1 nm, b = 1.7 ± 0.1 nm, α = 54 ± 2°, which are also summarized in Table S1. Total energy and energy per unit area for para-TTF/HA system and meta-TTF/HA system are summarized in Table S2 (Supporting information).
In combination with DFTcalculation, the detailed self-assembled structure based on the observed phenomena were understand, and the molecular models of the corresponding structures are shown in Figs. S5b and d (Supporting information), respectively. The calculated parameters are in good agreement with the experimental data. The para-TTF and meta-TTF molecules interact with the HA molecules through the O—H…N=C hydrogen bond in the selfassembly structure. For the different position of N atoms in the pyridine ring, the spatial conformation of these two co-assembled network is some different. In para-TTF/HA system, HA molecules are almost parallel to the skeleton of para-TTF. However, in meta-TTF/ HA system, there is a certain angle between HA molecules and meta-TTF skeleton. The total energy per unit area of para-TTF and meta-TTF is -0.346 kcal mol-1 Å-2 and -0.485 kcal mol-1 Å-2, respectively, indicating that meta-TTF/HA system is more stable.
In summary, the self-assembly behavior pyridine-TTF derivative para-TTF and meta-TTF without long alkyl chains has been detected for the first time. At the PO/HOPG interface, only para-TTF could form two kinds of linear structure, the compact arrangement and the loose arrangement. On the TCDB H-bonding networks, para-TTF and meta-TTF distrubed the H-bonding and induced the formation of new co-assembled structures. The difference is para-TTF located on top of two alkyl chains in the TCDB molecule. On the contrast, meta-TTF could form a grid nanostructure after introduction of TCDB network deformation. At the HA/HOPG interface, para-TTF and meta-TTF can form the stable linear self-assembly structures with HA molecules participating in the self-assembly structure. The self-assembled structures of TTF derivatives obtained by STM scanning showed that the packing of TTF cores can be effectively controlled by changing the substitution on the TTF molecule and the used solvent. This new phenomenon may enhance the understanding of the selfassembly of TTF at interfaces.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21472029, 21773041 and 21372136), Beijing National Laboratory for Molecular Sciences, the Ministry of Science and Technology of China (Nos. 2016YFA0200700 and 2017YFA0205001), Ningbo Natural Science Foundation (No. 2017A610013).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2018.09.019
| [1] |
M. Bendikov, F. Wudl, D.F. Perepichka, Chem. Rev. 104 (2004) 4891-4945. DOI:10.1021/cr030666m |
| [2] |
J.B. Sun, X.F. Lu, M. Ishikawa, et al., J. Mater. Chem. C 2 (2014) 8071-8076. DOI:10.1039/C4TC01362J |
| [3] |
D. Canevet, M. Salle, G.X. Zhang, D.Q. Zhang, D.B. Zhu, Chem. Commun. 17 (2009) 2245-2269. |
| [4] |
H. Jiang, X.J. Yang, Z.D. Cui, et al., Appl. Phys. Lett. 94 (2009) 3109785. |
| [5] |
B. Mukherjee, M. Mukherjee, Org. Electron. 12 (2011) 1980-1987. DOI:10.1016/j.orgel.2011.08.028 |
| [6] |
G. Yang, C.A. Di, G.X. Zhang, et al., Adv. Funct. Mater. 23 (2013) 1671-1676. DOI:10.1002/adfm.v23.13 |
| [7] |
Q.Y. Zhu, Y. Liu, W. Lu, et al., Inorg. Chem. 46 (2007) 10065-10070. DOI:10.1021/ic700672e |
| [8] |
W. Pan, X.W. Xiao, Z.Q. Wang, et al., Synthetic Met. 194 (2014) 132-136. DOI:10.1016/j.synthmet.2014.04.021 |
| [9] |
Z.N. Yin, Y.H. Li, Y.G. Sun, et al., Inorg. Chem. 55 (2016) 9154-9157. DOI:10.1021/acs.inorgchem.6b01632 |
| [10] |
C.Y. Jia, S.X. Liu, C. Ambrus, et al., Inorg. Chem. 45 (2006) 3152-3154. DOI:10.1021/ic060056f |
| [11] |
R. Bertani, P. Sgarbossa, A. Venzo, et al., Coordin. Chem. Rev. 254 (2010) 677-695. DOI:10.1016/j.ccr.2009.09.035 |
| [12] |
H. Wang, H.J. Bao, S.H. Shen, et al., Inorg. Chim. Acta 474 (2018) 164-169. DOI:10.1016/j.ica.2018.02.010 |
| [13] |
M. Nakaya, M. Shikishima, M. Shibuta, et al., ACS Nano 6 (2012) 8728-8734. DOI:10.1021/nn302405r |
| [14] |
S.C. Lee, A. Ueda, H. Kamo, et al., Chem. Commun. 48 (2012) 8673-8675. DOI:10.1039/c2cc34296k |
| [15] |
M.N. Nair, C. Mattioli, M. Cranney, et al., J. Phys. Chem. C 119 (2015) 9334-9341. DOI:10.1021/acs.jpcc.5b00857 |
| [16] |
M.M.S. Abdel-Mottaleb, E. Gomar-Nadal, S. De Feyter, et al., Nano Lett. 3 (2003) 1375-1378. DOI:10.1021/nl034515a |
| [17] |
S.L. Lee, Y.C. Chu, H.J. Wu, C.H. Chen, Langmuir 28 (2012) 382-388. DOI:10.1021/la203148h |
| [18] |
M.M.S. Abdel-Mottaleb, E. Gomar-Nadal, M. Surin, et al., J. Mater. Chem. 15 (2005) 4601-4615. DOI:10.1039/b509336h |
| [19] |
J. Xu, X.W. Xiao, K. Deng, Q.D. Zeng, Nanoscale 8 (2016) 1652-1657. DOI:10.1039/C5NR07345F |
| [20] |
Y.T. Shen, L.J. Zeng, D. Lei, et al., J. Mater. Chem. 21 (2011) 8787-8791. DOI:10.1039/c1jm10260e |
| [21] |
J. Liu, T. Chen, X. Deng, et al., J. Am. Chem. Soc. 133 (2011) 21010-21015. DOI:10.1021/ja209469d |
| [22] |
X.H. Kong, K. Deng, Y.L. Yang, Q.D. Zeng, C. Wang, J. Phys. Chem. C 111 (2007) 17382-17387. DOI:10.1021/jp074682p |
| [23] |
X.M. Zhang, Y.T. Shen, S. Wang, et al., Sci. Rep. 2 (2012) 00742. DOI:10.1038/srep00742 |