 2019, Vol. 30
2019, Vol. 30 


Alkenylation of aromatics with alkynes has been considered as a direct and efficient method for synthesizing alkenylaromatics, which serve as many industrially important intermediates for producing pharmaceuticals, agrochemicals, natural products, flavors and dyes [1-3]. Owing to its low-cost and high activity, the acid catalyzed process shows great potential for the synthesis of alkenylaromatics [4-8]. Catalysis by solid acid presents a promising strategy for diverse transformations in terms of its feature of clean, easy-separation, catalyst reusability, and its applicability toward continuous production in an industrial scale.
In contrast to well-established alkylation, the acid-catalyzed alkenylation still remains a great challenge. The biggest issue is to efficiently overcome the heavy oligomerization of alkynes, owing to the poor stability of vinyl cation intermediate species [9]. The regular pore channels, a large amount of acidic sites, corrosionresistance, and environmentally benign feature of zeolites endow them with great attention in alkylation in academic and industrial fields [10-13]. However, the serious coke deposition due to the narrow pore channels of zeolites and the stability of vinyl cation species depresses the broad use of zeolites as solid acid catalysts in alkenylation [14, 15]. Therefore, rare report on alkenylation over zeolite can be found.
From reference, Sartori did a pioneering work on alkenylation of aromatics over HSZ-360 zeolite [16]. Unfortunately, the irreconcilable contradiction between the selectivity and catalytic activity led to an unsatisfactory reaction. The zeolite calcined at lower temperature exhibited high catalytic activity, but the considerable amount of acetophenone (5%–20%) was detected; the higher calcination temperature could efficiently compress the formation of acetophenone, but resulted in remarkable decrease in catalytic activity. Moreover, the enlargement in scope of substrates for alkenylation over zeolite is also required. Recently, it was demonstrated that the Fe-containing mesoporous alluminosilicate exhibited vey high activity for alkenylation of phenols with arylsubstituted alkynes under mild conditions [17]. However, the further increase in selectivity is indispensable. Therefore, the development of novel and robust solid acid catalysts for alkenylation is highly desirable, but it still remains a great challenge.
Owing to its strong acidity, the heteropolyacid (HPA) acted as a very efficient solid acid catalyst for alkylation. Among them, PTA is usually employed as a good catalyst for its high acidic strength and relatively high thermal stability [18, 19]. However, the lower surface area (about 5–8 m2/g) limits its application as a heterogeneous catalyst. Loading HPA on an appropriate carrier with high specific surface area and pore volume can be an efficient approach to address this issue [20-24]. It was previously demonstrated that the supported PTA catalyst on MCM-41 under optimized conditions exhibited good catalytic performance in alkenylation [14]. However, further improvement in catalytic activity, selectivity and the expansion in reactants scope are required.
From the previous report in our Advanced Catalytic Materials (ACM) research group, owing to high PTA dispersity and facilitated mass transfer by the short mesoporous channels of the spherical morphology, the supported PTA catalyst on monodisperse mesoporous silica nanosphere (MSN) demonstrated much higher catalytic activity and stability for alkenylation and the other diverse transformations in comparison with traditional mesoporous silica carriers like MCM-41. PTA/MSN has been considered as a promising practical solid acid catalyst for diverse reactions [25]. However, the further increase in catalytic performance is required.
Owing to the influence of unique morphology on the diffusion length of the reactants and products, exposure degree of active sites, dispersity, crystalline phase, and so on, the supported-type catalyst on spherical hollow mesoporous silica demonstrated much superior catalytic performance in many transformation processes [26-33]. In this work, the hollow mesoporous silica submicrosphere with high surface area (902 m2/g) and large mesopore pore volume (1.31 cm3/g) was prepared through a facile and scalable soft-hard dual-template assisted sol-gel approach. In comparison with the previously reported papers [34-39], in this process, the size-preselected glucose-derived carbon submicroparticle was used to replace ploymer sphere, and no extra precious additives like n-octadecyltrimethoxysilane (C18TMS), as well as the hollow mesoporous silica submicrosphere with both higher surface area and larger mesopore volume were achieved. The supported PTA on SHMS demonstrates much higher catalytic activity than PTA/SMS for diverse reactions including alkenylation, esterification, alkylation, and benzylation, and also exhibits outstanding catalytic performance for the diverse α-arylstyrenes via solid acid-mediated alkenylation. Various characterization techiniques including TEM, N2 adsorption–desorption, XRD, and NH3-TPD were used to characterize the nature of SHMS and SMS carriers and their corresponding supported PTA catalysts, and also to reveal the structure-performance relationship. PTA/SHMS could be considered as a practical solid acid catalyst for diverse transformations.
In a typical experiment, the carbon submicrospheres with preselected particle size were prepared by a modified hydrothermal process of glucose followed by step-by-step centrifugation process [34]. Typically, 5 g of glucose were dissolved in 80 mL deioned water under stirring at room temperature to form a clear solution, and then the resulting aqueous solution was transferred into a Teflon-lined autoclave (100 mL) for the hydrothermal carbonization process at 170 ℃ for 10 h to form carbon nano/micro-particles under autogeneous pressure. After that, the larger particles were removed by centrifugalizing the upper black liquid of the above resulting mixture by hydrothermal process at 3000 rpm. Subsequently, the size preselected samples from further centrifugation at 10, 000 rpm were purified by repeatedly washing them in water and ethanol for three times, followed by drying at 60 ℃ for 6 h. The spherical hollow mesoporous silica was prepared by a facile and scalable two-step soft-hard dual-template assisted solgel approach by using the as-synthesized size-preselected NMCP as a hard template and CTAB as a soft template, respectively. The as-synthesized NMCP (0.2 g) were re-dispersed in a mixed solution of ethanol and water by stirring and ultrasonic treatment, and then 570mL NH3·H2O and 0.28 g CTAB were added into the above solution. After vigorously stirring for 1 h, 400 μL TEOS was dropwisely added into the dispersion liquid with continuous stirring for 6 h. The products were collected by filtration and washed with water and ethanol, followed by drying at 80 ℃ for 5 h. Finally, the SHMS was prepared by calcining the sample at 550 ℃ for 6 h to remove both soft and hard templates. SMS was synthesized by the previously reported method [25]. Typically, the synthesis was performed under mild conditions with a typical composition of 1.0 TEOS:0.064 CTAB:2.5 urea:300 H2O at a molar basis. 0.816 g CTAB and the required amount of urea were dissolved in 200 g H2O and stirred for 1 h, and then 7.3 g of TEOS was added dropwisely into the above solution. The resulting mixture was stirred at 80 ℃ for 2 h. After that, the milky liquid was transferred into Teflon-lined autoclave and heated at 100 ℃ for 20–60 h. The obtained solid product was recovered, washed, and then dried at 105 ℃ for 20 h. The final SMS was obtained by calcining the above samples at 550 ℃ for 6 h. The supported PTA catalysts containing PTA/SHMS and PTA/SMS with 25 wt% of PTA loading were prepared by our previously reported vacuum assisted wet impregnation with heating method (IMPVH) [14]. Typically, SHMS (1.0 g) was merged into an aqueous solution of PTA (according to previous reports [25], Keggin structure of PTA, the concentrations is 0.36 g/mL) in a vacuum environment with heating at 128 ℃. After that, the impregnated samples were dried at 105 ℃ in air overnight, followed by calcinations in air at 300 ℃ for 3 h, and the PTA/SHMS catalyst with a loading of 25 wt% were obtained. By using same process, the PTA/SMS catalyst was prepared for comparison [25].
The morphology and textural properties of the as-formed supports and the supported PTA catalysts were characterized by Transmission electron microscopy (TEM) and nitrogen adsorptiondesorption experiments. TEM images were obtained by using a Tecnai F30 HRTEM instrument (FEI Corp.) at an acceleration voltage of 300 kV. Nitrogen adsorption experiments at -196 ℃ were carried out on a Beishide 3H-2000PS1 instrument to measure the surface area and pore volume, and the samples were degassed at 130 ℃ for 10 h prior to the N2 adsorption experiment.
The crystalline structure and acidic properties of the supported PTA catalysts on the as-formed SHMS and SMS silica spheres were measured by X-ray diffraction (XRD) and NH3 temperature programmed desorption (NH3-TPD) techniques. XRD profiles were collected from 10° to 80° at a step width of 0.02° using Rigaku Automatic X-ray Diffractometer (D/Max 2400) equipped with a CuKα source (λ = 1.5406 Å). NH3-TPD measurements were performed on a Builder Chemisorption (PCA-1200) instrument with a thermal conductivity detector (TCD) to measure the desorbed NH3. After pre-treatment of 50 mg samples in Ar (up to 300 ℃ with a ramp rate of 10 ℃/min, and then kept for 0.5 h under 30 mL/min Ar flow), the samples were saturated with ammonia (10% NH3-90% Ar) at 10 ℃ via the pulse injection of ammonia in an Ar stream. The desorption step was carried out from 100 ℃ to 700 ℃ at a ramp rate of 10 ℃/min with an Ar flow rate of 30 mL/min. The NH3-TPD profiles were obtained via monitoring the desorbed ammonia with a thermal conductivity detector (TCD).
The catalytic performance of the as-synthesized containing PTA/SHMS and PTA/SMS for direct alkenylation of aromatics with phenylacetylene was on a stainless steel autoclave reactor. In addition to the desired amount of catalyst, 15 g reactant mixture containing aromatics and phenylacetylene was added into autoclave reactor. After that the autoclave was purged with N2 for three times, and pre-filled with N2 upto 0.7–0.75 MPa to maintain 1.0 MPa of reaction pressure at reaction temperature. The reaction mixture got a homogeneous state after stirring 30 min at room temperature, then was heated up to the desired reaction temperature, and start to a required reaction time. After the reaction process, the mixture was quickly cooled down to room temperature and then filtered for catalyst separation. Quantitative analysis of the collected product was performed on a FULI 9790 Ⅱ GC equipped with HP-5 column, 30 m × 0.32 mm × 0.25 mm, and FID detector. 1H NMR experiments were performed for the structure identification of samples (see Supporting information). The phenylacetylene conversion was calculated by weight percent of the consumed phenylacetylene in the total phenylacetylene amount added; the selectivity to α-arylstyrene was calculated by weight percent of desired α-arylstyrene in total products. The similar process was performed for synthesizing diverse α- arylstyrenes via solid acid-mediated alkenylation of diverse aromatics as substrates. The other solid acid catalyzed transformations were performed in a three-neck flask, and the detailed reaction conditions are as follows: a) For esterification reaction, acetic acid 100 mmol, ethanol 1 mol, catalyst dosage 50 mg, reaction temperature 70 ℃, reaction time 5 h. b) For alkylation reaction, styrene 100 mmol, benzene 500 mmol, catalyst dosage 50 mg, reaction temperature 70 ℃, reaction time 5 h. c) For benzylation, benzyl alcohol 50 mmol, benzene 125 mmol, catalyst dosage 200 mg, reaction temperature 110 ℃, reaction time 5 h. The reaction rate was calculated on the basis of molar amount of transformed reactants per gram catalyst per hour.
TEM experiments were performed to obtain the morphology and size of the as-formed SHMS and SMS silica spheres. Fig. 1 presents the typical TEM images of the as-synthesized, and their particle size is depicted in Table S1 (Supporting information). Figs. 1a and b clearly demonstrate that the spherical hollow mesoporous silica with 80–100 nm of cavity size and about 30 nm shell thickness was successfully synthesized by the a facile and scalable two-step soft-hard dual-template assisted sol-gel approach by using the as-synthesized size-preselected NMCP as a hard template and CTAB as a soft template. The SHMS submicrospheres feature the rough outer surface. For comparison, the spherical solid mesoporous silica (SMS) with about 114 nm (113 ± 4 nm) particle size was also prepared (Figs. 1c and d). From Fig. 1 and Table S1, SHMS definitely exhibits a larger particle size than SMS.

|
Download:
|
| Fig. 1. (a–d) TEM images of the as-synthesized SHMS (a, b) and SMS (c, d) mesoporous silica submicrospheres. (e) Schematic illustration for the formation of SHMS with high surface area and large mesopore volume via a facile and scalable OSDSG method. | |
Fig. 1e illustrates the preparation process of SHMS. The sizepreselected carbon nano/micro-spheres were prepared by hydrothermal carbonization of glucose with the subsequently controlled centrifugation process. The carbon spheres were used as the hard template to prepare SHMS without further modification. From references [40, 41], the large amount of silicates are quickly formed under a high alkali concentration, which can wrap several surfactant micelles. Then the worm-like silicated micelles are formed. The worm-like-silicated-micelles-containing aggregates are produced on the carbon sphere surface. After removing both soft and hard template through subsequent calcination process, SHMS submicrospheres with cavities were successfully synthesized.
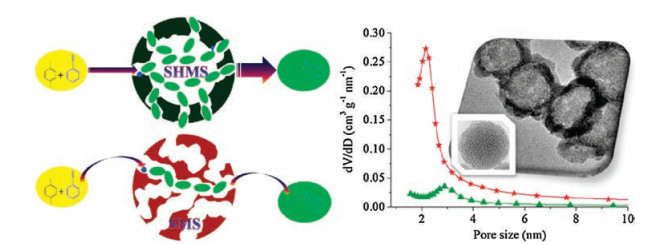
Nitrogen adsorption-desorption isotherms of the as-synthesized SHMS and SMS are presented in Fig. 2a, and the BJH pore size distribution from adsorption is shown as inset in Fig. 2a. The textural properties of both of the two silica nanospheres are listed in Table S1. From Fig. 2a, all of the isotherms feature a typical type Ⅳ characteristic according to IUPAC classification and exhibit H1 hysteresis with a featured capillary condensation in mesopores, indicating the existence of mesopore [42, 43]. From Fig. 3, the quick rising adsorption at a quite low P/P0 on the isotherms of the assynthesized hollow mesoporous silica spheres suggests the existence of micropores, which is a result of removing CTAB tails trapped into the silica structure [35]. It is interesting that the isotherms present two clear capillary condensation steps: one is at 0.15–0.60 of a lower relative pressure (p/p0) and the other is at 0.7– 1.0 of a higher relative pressure region, implying the feature of bimodal pores [44-47]. The former features the formation of mesopores in both of the two samples, and the latter shows the existence of cavities. From the inset in Fig. 2, the SHMS and SMS exhibit two types of pores containing both mesopores (the centre of pore distribution from the adsorption branch is 2.1 nm and 2.9 nm towards SHMS and SMS, respectively) and macropores (50– 200 nm, based on IUPAC classification). The mesopores are generated by removing the templating surfactant, whereas the macropores can be originated from the removal of carbon hard template for SHMS and the accumulation of silica sphere for SMS. SHMS shows a smaller pore size of mesopores than SMS (2.1 nm vs. 2.9 nm). From Table S1, SHMS exhibits higher specific surface area towards total and larger pore volume for both of micropores and mesopores than SMS.

|
Download:
|
| Fig. 2. (a) N2 adsorption-desorption isotherms of the as-synthesized SHMS and SMS carriers and the supported PTA solid acid catalysts. Inset is the Barrett-Joyner-Halenda (BJH) pore size distribution from adsorption branch. (b, c) XRD patterns (b) and NH3-TPD profiles (c) of the as-prepared PTA/SHMS and PTA/SMS catalysts with 25 wt% of PTA loading. | |

|
Download:
|
| Fig. 3. Schematic illustration for improved catalytic performance over PTA/SHMS solid acid catalyst in contrast to PTA/SMS. | |
The supported PTA solid acid catalysts on mesoporous silica supports have attracted great attention in diverse transformations owing their excellent catalytic performance [48-52]. By employing the as-synthesized SHMS and SMS as carriers, the supported PTA solid acid catalysts with 25 wt% of PTA loading (PTA/SMSN and PTA/WMSN) were prepared via the previously reported impregnation method [14]. The N2 adsorption desorption isotherms, XRD patterns, and NH3-TPD profiles are presented in Fig. 2. From Fig. 2a and Table S1, both PTA/SHMS and PTA/SMS catalysts exhibit a lowering specific area and a decreasing pore volume in contrast to their corresponding carriers, which is consistent with those in previous reports [53-55]. More interestingly, the decreasing degree of specific surface area and pore volume of PTA/SMS after supporting PTA is much larger than that of PTA/SHMS (almost no change in volume and pore diameter for the as-synthesized PTA/ SHMS can be observed). Thus would be ascribed to the higher dispersity of PTA on SHMS than that on SMS, depending on their different morphologies. The higher PTA dispersity of PTA/SHMS than that of PTA/SMS can be affirmed by the XRD patterns (Fig. 2b). From Fig. 2b, the XRD characteristic peaks corresponding to PTA on PTA/SHMS can be hardly seen, implying the high PTA dispersity by the high surface and large pore volume of SHMS [14, 56-58]. Furthermore, the acidic properties of the two supported PTA catalysts were investigated, and Fig. 2c presents the NH3-TPD profiles. From Fig. 2c and quantitative result, PTA/SHMS shows much higher amount of acidic sites (0.56 mmol/g) in contrast to PTA/SMS (0.26 mmol/g), ascribed to its larger exposure degree of acidic sites, higher surface area, larger pore volume, and the resulting higher PTA dispersity in comparison with PTA/SMS, those are strongly dependent on its unique hollow mesoporous morphology.
Employing alkenylation, esterification, alkylation, and benzylation as acidic mediated model reactions, the catalytic performance of PTA/SHMS and PTA/SMS catalysts were investigated, and the reaction rate results are presented in Table 1. From Table 1, in comparison with the blank experiments, both PTA/SHMS and PTA/ SMS catalysts demonstrate significant catalysis in the diverse solid acid catalyzed transformations. Although SHMS shows much higher particle diameter (outer diameter, larger than 150 nm) than SMS (about 113 nm), the PTA/SHMS catalyst shows much higher reaction rate for the diverse reactions than PTA/SMS, ascribed to the unique hollow mesoporous sphere morphology and their resulting high surface area and pore volume lead to high PTA dispersity, which leads to high amount of acidic sites, besides the strengthened mass transfer by hollow structure [26-33]. However, it can be found that the increasing degree in activity is less than the one in the amount of acidic sites (0.56 mmol/g vs. 0.26 mmol/g), which might be the smaller mesopore size of PTA/SHMS in contrast to PTA/SMS.
|
|
Table 1 Reaction rate for diverse model reactions over the different solid acid catalysts. |
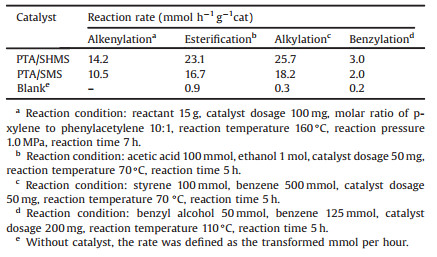
Fig. 3 illustrates the enhancing effect of hollow mesoporous morphology of PTA/SHMS. Owing to the hollow morphology of PTA/ SHMS, the diffusion of reactants, intermediates, and products can be efficiently enhanced by the shortening diffusion length [26-33]. As a result, thePTA/SHMS solid acid catalyst demonstrates much superior catalytic activity in comparison with PTA/SMS. From the reaction results listed in Table 1, the lower degree in the increasing activity than that in the increasing acidic sites can be the weakened mass transfer from the smaller mesopore size of PTA/SHMS, although the shortened diffusion distance by hollow structure strengthens mass transfer. Therefore, in order to further improve the catalytic performance of supported PTA catalyst, the modulation of mesopore size of hollow mesoporous silica spheres would be addressed in the future work. This approach by shorting length of mesopore of the mesoporous solid acid catalysts can be considered as a sapiential strategy for improving reaction rate of solid acid catalysts for diverse transformations, besides the increasing acidic sites resulting from the enlarged surface area and pore volume by the unique hollow mesoporous morphology. The catalytic performance of PTA/SHMS catalyst can be further modulated and optimized through adjusting the size of cavity, shell thickness, and also the size and structure of mesopores within the mesoporous shells of SHMS.
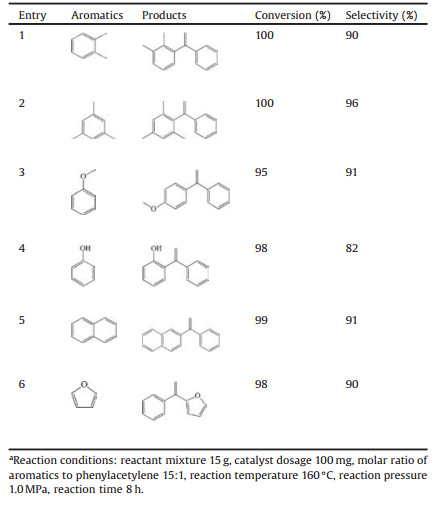
Moreover, the catalytic cycle performance of PTA/SHMS catalyst was investigated, and the results are preented in Table S2 (Supporting information). The results shows that no visible loss in catalytic performance on the devloped PTA/SHMS catalyst can be observed, suggesting the outstanding catalytic stability. Furthermore, the developed PTA/MSN-0.025 in this work could be a promising solid acid catalyst for the production of alpha-arylstyrenes through direct alkenylation of diverse aromatics with phenylacetylene. Herein, the scope of substrates for the alkenylation over the developed PTA/ SHMS catalyst was investigated, and the reaction results are listed in Table 2. The molecular structures of alkenylated products are identified by 1HNMR (Supporting information). From Table 2, the developed solid acid catalyst demonstrates excellent catalytic performance with more 95% conversion with high selectivity for the alkenylation of diverse aromatics, suggesting the broad scope of aromatics for the alkenylation for production of diverse α- arylstyrenes over the developed solid acid catalyst.
|
|
Table 2 Summary of the alkenylation of diverse aromatics with phenylacetylene to their corresponding α-arylstyrenes over the developed supported PTA solid acid catalyst on SHMS.a |
{kind=link}
{kind=link}
{kind=link}
In summary, the supported PTA solid acids on the two spherical mesoporous silica carriers including SHMS and SMS were prepared and evaluated for diverse solid acid-mediated transformations. PTA/SHMS catalyst demonstrates much superior catalytic performance for the investigated diverse transformations than PTA/SMS, ascribed to the more acidic sites and intensified mass transfer by the unique hollow mesoporous morphology. This work also shows that the modulation of diffusion distance through morphology adjustment of mesoporous silica carriers can be considered as a sapiential strategy for preparing the outstanding solid acid catalysts. The PTA/SHMS also shows promising catalytic properties for alkenylation of diverse aromatics. Moreover, the size-preselected glucose-derived carbon nano/micro-spheres were used as hard template, which presents a facile method for controlling the size of cavities of diverse spherical hollow materials.
AcknowledgmentsThis work is financially supported by the National Natural Science Foundation of China (Nos. 21276041 and U1610104) and by the Chinese Ministry of Education via the Program for New Century Excellent Talents in University (No. NCET-12-0079).
Appendix A. Supplementary dataSupplementary materialrelated tothis article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.08.014.
| [1] |
N.P. Grimster, C. Gauntlett, C.R. Godfrey, M.J. Gaunt, Angew. Chem. Int. Ed. 117 (2005) 3185-3189. |
| [2] |
Y. Yamamoto, Chem. Soc. Rev. 43 (2014) 1575-1600. DOI:10.1039/C3CS60369E |
| [3] |
C. Jia, D. Piao, J. Oyamada, et al., Science 287 (2000) 1992-1995. DOI:10.1126/science.287.5460.1992 |
| [4] |
K. Komeyama, R. Igawa, K. Takaki, Chem. Commun. 46 (2010) 1748-1750. DOI:10.1039/b920695g |
| [5] |
R. Li, S.R. Wang, W.J. Lu, Org. Lett. 9 (2007) 2219-2222. DOI:10.1021/ol070737u |
| [6] |
C.E. Song, D. Jung, S.Y. Choung, E.J. Roh, S. Lee, Angew. Chem. Int. Ed. 43 (2004) 6183-6185. |
| [7] |
D.S. Choi, J.H. Kim, U.S. Shin, R.R. Deshmukh, C.E. Song, Chem. Commun. (2007) 3482-3484. |
| [8] |
S.A. Aristov, A.V. Vasil'ev, G.K. Fukin, A.P. Rudenko, Russ. J. Org. Chem. 43 (2007) 691-705. DOI:10.1134/S1070428007050107 |
| [9] |
J.A. Reilly, J.A. Nieuwland, J. Am. Chem. Soc. 50 (1928) 2564-2566. DOI:10.1021/ja01396a037 |
| [10] |
B.O. Dalla Costa, C. Querini, Appl. Catal. A 385 (2010) 144-152. DOI:10.1016/j.apcata.2010.07.007 |
| [11] |
K. Liu, S. Xie, S. Liu, et al., J. Catal. 283 (2011) 68-74. DOI:10.1016/j.jcat.2011.07.004 |
| [12] |
M. Milina, S. Mitchell, Z.D. Trinidad, D. Verboekend, J. Pérez-Ramírez, Catal. Sci. Technol. 2 (2012) 759-766. DOI:10.1039/c2cy00456a |
| [13] |
G. Kostrab, M. Lovic, I. Janotka, M. Bajus, D. Mravec, Appl. Catal. A 335 (2008) 74-79. DOI:10.1016/j.apcata.2007.11.019 |
| [14] |
Z. Zhao, Y. Dai, T. Bao, R. Li, G. Wang, J. Catal. 288 (2012) 44-53. DOI:10.1016/j.jcat.2011.12.024 |
| [15] |
Z. Zhao, R. Jin, X. Lin, G. Wang, Energy Sources Part A 35 (2013) 1761-1769. DOI:10.1080/15567036.2010.531503 |
| [16] |
G. Sartori, A. Pastorío, C. Porta, et al., Tetrahedron Lett. 36 (1995) 9177-9180. DOI:10.1016/0040-4039(95)01934-A |
| [17] |
S. Haldar, S. Koner, Beilstein J. Org. Chem. 9 (2013) 49-55. DOI:10.3762/bjoc.9.6 |
| [18] |
W. Ninomiya, M. Sadakane, S. Matsuoka, H. Naitou, W. Ueda, Green Chem. 11 (2009) 1666-1674. DOI:10.1039/b914515j |
| [19] |
K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya, A. Satsuma, Green Chem. 11 (2009) 1627-1632. DOI:10.1039/b913737h |
| [20] |
W. Luo, T. Zhao, L. Wang, et al., J. Am. Chem. Soc. 138 (2016) 12586-12595. DOI:10.1021/jacs.6b07355 |
| [21] |
A. Patel, S. Singh, Micropor. Mesopor. Mater. 195 (2014) 240-249. DOI:10.1016/j.micromeso.2014.04.039 |
| [22] |
T. Zhao, W. Luo, Y. Deng, et al., Nano Energy 26 (2016) 16-25. DOI:10.1016/j.nanoen.2016.04.050 |
| [23] |
T. Kumaraguru, A.V. Devi, V. Siddaiah, K. Rajdeo, N.W. Fadnavis, Appl. Catal. A 486 (2014) 55-61. DOI:10.1016/j.apcata.2014.08.026 |
| [24] |
G. Sunita, B.M. Devassy, A. Vinu, et al., Catal. Commun. 9 (2008) 696-702. DOI:10.1016/j.catcom.2007.08.007 |
| [25] |
Z.K. Zhao, X.H. Wang, Y.H. Jiao, et al., RSC Adv. 6 (2016) 9072-9081. DOI:10.1039/C5RA26432D |
| [26] |
W. Tang, X. Wu, Y. Chen, Mater. Lett. 168 (2016) 111-115. DOI:10.1016/j.matlet.2015.12.142 |
| [27] |
X. Yang, S. Liao, J. Zeng, Z. Liang, Appl. Surf. Sci. 257 (2011) 4472-4477. DOI:10.1016/j.apsusc.2010.12.096 |
| [28] |
X. Lin, S. Zhao, L. Fu, et al., Mol. Catal. 437 (2017) 18-25. DOI:10.1016/j.mcat.2017.04.026 |
| [29] |
J. Li, Y. Xu, D. Wu, Y. Sun, Catal. Today 148 (2009) 148-152. DOI:10.1016/j.cattod.2009.02.046 |
| [30] |
T. Umegaki, M. Hoshino, Y. Watanuki, Y. Kojima, Micropor. Mesopor. Mater. 223 (2016) 152-156. DOI:10.1016/j.micromeso.2015.11.010 |
| [31] |
Y. Yin, M. Chen, S. Zhou, L. Wu, J. Mater. Chem. 22 (2012) 11245-11251. DOI:10.1039/c2jm31138k |
| [32] |
Y. Li, B.P. Bastakoti, H. Abe, et al., RSC Adv. 5 (2015) 97928-97933. DOI:10.1039/C5RA17340J |
| [33] |
J. Wang, C. Liu, I. Hussain, et al., RSC Adv. 6 (2016) 54623-54635. DOI:10.1039/C6RA08501F |
| [34] |
R. Liu, C. Wang, Ceramics Int. l 41 (2015) 1101-1106. DOI:10.1016/j.ceramint.2014.09.035 |
| [35] |
S. Ghasemi, Z.J. Farsangi, A. Beitollahi, et al., Ceramics Int. 43 (2017) 11225-11232. DOI:10.1016/j.ceramint.2017.05.172 |
| [36] |
H. Zhang, H. Xu, M. Wu, et al., J. Mater. Chem. B 3 (2015) 6480-6489. DOI:10.1039/C5TB00634A |
| [37] |
Z.J. Farsangi, A. Beitollahi, B.D. Hatton, et al., RSC Adv. 6 (2016) 67592-67598. DOI:10.1039/C6RA10856C |
| [38] |
W. Zhao, M. Lang, Y. Li, L. Li, J. Shi, J. Mater. Chem. 19 (2009) 2778-2783. DOI:10.1039/b822444g |
| [39] |
F. Wang, Y. Tang, B. Zhang, B. Chen, Y. Wang, J. Colloid Int. Sci. 386 (2012) 129-134. DOI:10.1016/j.jcis.2012.06.088 |
| [40] |
Y. Lin, C. Tsai, Y. Huang, et al., Chem. Mater. 17 (2005) 4570-4573. DOI:10.1021/cm051014c |
| [41] |
K. Zhang, L. Xu, J. Jiang, et al., J. Am. Chem. Soc. 135 (2013) 2427-243. DOI:10.1021/ja3116873 |
| [42] |
Z.K. Zhao, Y.T. Dai, J.H. Lin, G.R. Wang, Chem. Mater. 26 (2014) 3151-3161. DOI:10.1021/cm5005664 |
| [43] |
Z.K. Zhao, J.F. Ran, Y.L. Guo, B.Y. Miao, G.R. Wang, Chin. J. Catal. 37 (2016) 1303-1313. DOI:10.1016/S1872-2067(15)61118-4 |
| [44] |
N. Zucchetto, M.J. Reber, L. Pestalozzi, et al., Micropor. Mesopor. Mater. 257 (2018) 232-240. DOI:10.1016/j.micromeso.2017.08.046 |
| [45] |
H. Liu, H. Liu, J. Mater. Chem. A 5 (2017) 9156-9162. DOI:10.1039/C7TA01255A |
| [46] |
C. Weinberger, X. Cao, M. Tiemann, J. Mater. Chem. A 4 (2016) 18426-18431. DOI:10.1039/C6TA07772B |
| [47] |
D. Feng, Z. Lin, M. Liu, et al., Micropor. Mesopor. Mater. 222 (2016) 138-144. DOI:10.1016/j.micromeso.2015.10.014 |
| [48] |
D.P. Sawant, A. Vinu, F. Lefebvre, S.B. Halligudi, J. Mol. Catal. A 262 (2007) 98-108. DOI:10.1016/j.molcata.2006.08.027 |
| [49] |
P. Kamala, A. Pandurangan, Catal. Commun. 9 (2008) 2231-2235. DOI:10.1016/j.catcom.2008.05.003 |
| [50] |
Z.K. Zhao, X.H. Wang, Appl. Catal. A 526 (2016) 139-146. DOI:10.1016/j.apcata.2016.08.028 |
| [51] |
F.N.D.C. Gomes, F.M.T. Mendes, M.M.V.M. Souza, Catal. Today 279 (2017) 296-304. DOI:10.1016/j.cattod.2016.02.018 |
| [52] |
T.H. Kang, J.H. Choi, Y. Bang, et al., J. Mol. Catal. A 396 (2015) 282-289. DOI:10.1016/j.molcata.2014.10.015 |
| [53] |
Z.K. Zhao, X.H. Wang, Appl. Catal. A 503 (2015) 103-110. DOI:10.1016/j.apcata.2015.07.007 |
| [54] |
S.S. Sakate, S.B. Kamble, R.C. Chikate, C.V. Rode, New J. Chem. 41 (2017) 4943-4949. DOI:10.1039/C7NJ00375G |
| [55] |
M. Almohalla, I. Rodríguez-Ramos, A. Guerrero-Ruiz, Catal. Sci. Technol. 7 (2017) 1892-1901. DOI:10.1039/C7CY00155J |
| [56] |
Y. Shen, X.H. Lu, C.C. Wei, et al., Mol. Catal. 433 (2017) 185-192. DOI:10.1016/j.mcat.2016.12.012 |
| [57] |
L. Frattini, M.A. Isaacs, C.M.A. Parlett, et al., Appl. Catal. B 200 (2017) 10-18. DOI:10.1016/j.apcatb.2016.06.064 |
| [58] |
M. Srinivas, G. Raveendra, G. Parameswaram, P.S. Prasad, N. Lingaiah, J. Mol. Catal. A 413 (2016) 7-14. DOI:10.1016/j.molcata.2015.10.005 |