 2019, Vol. 30
2019, Vol. 30 


b Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200032, China;
c Shanghai Research Institute of Chemical Industry Co., LTD., Shanghai 200062, China
Due to its strong electron-withdrawing effects (Hammett constants σp = 0.50, σm = 0.40) and high lipophilicity (Hansch parameter π = 1.44) [1-3], trifluoromethylthio group (CF3S) has been of increasing importance in drug design and development. CF3S-containing pharmaceuticals such as Tiflorex, Cefazaflur and Toltrazuril have been continuously developed [4]. Therefore, significant efforts have been directed towards the development of efficient methods for the incorporation of a CF3S group into organic molecules [4-14]. Compared with traditional methods such as halogen-fluorine exchange [15, 16] and trifluoromethylation of sulfur compounds [17, 18], direct trifluoromethylthiolation is obviously an efficient and straightforward strategy for CF3S incorporation [4]. A variety of direct trifluoromethylthiolation approaches have been developed, including electrophilic, radical and nucleophilic trifluoromethylthiolation (Scheme 1, reaction (1)) [19-25]. In these approaches, the synthesis of CF3S-containing reagents is usually required, and the reagents may be expensive, sensitive to air or difficult to prepare. Interestingly, the classic trifluoromethylation reagent, TMSCF3, could also enable the trifluoromethylthiolation in the presence of a sulfur source (reaction (1)) [26-28]. But the trifluoromethylation reagent is quite volatile. Apparently, the development of effective trifluoromethylthiolation protocols by using easily handled reagents is highly desirable.

|
Download:
|
| Scheme 1. Trifluoromethylthiolation strategies. | |
As a versatile intermediate, difluorocarbene has found widespread application in a wide range of reactions, such as [2 + 1] cyclization and X-H insertion (X = N, O, S, etc.) [29, 30]. Difluorocarbene is quite electrophilic and thus would be readily trapped by surrounding nucleophiles to generate unavoidable by-products. The side reactions are usually ignored and the by-products would therefore be discarded. However, the development of these unwanted processes into synthetic tools may provide a foundation for the exploration of difluorocarbene chemistry.
Ph3P+CF2CO2- (PDFA), a reagent which was developed by our group [31-40] and was also used by other groups [41-46], could be easily prepared via a reaction of Ph3P with BrCF2CO2K. Pure product could be obtained simply by washing, and the reagent is bench-stable and easy to handle. We have previously found that PDFA could act as an efficient difluorocarbene reagent to realize trifluoromethylthiolation (Scheme 1, reaction (2)) [36, 38]. Since an external fluoride anion was used to install the CF3S moiety and the trifluoromethylthiolation reaction occurred very rapidly (5 min or 20 min), 18F-trifluoromethylthiolation was examined and successfully achieved [36, 38]. Based on our previous observation that side reaction of difluorocarbene would produce a fluoride anion [34],
we have now investigated trifluoromethylthiolation by using this difluorocarbene reagent without the presence of an external fluoride anion (Scheme 1, reaction (3)). The generation of fluoride anion from difluorocarbene, a process which has always been considered as a side reaction, was an important chemical step in this protocol. Interestingly, although the trifluoromethylthiolation reaction involved multi-sequential steps, the cleavage of C–F bond, the formation of CF2=S bond, F-C(S)F2 bond, and C-SCF3 bond, the conversion proceeded fast and was completed within 10 min.
As our previous work has shown that DBU [1, 8-diazabicyclo [5.4.0]undec-7-ene] could well promote the generation of fluoride anion from difluorocarbene in DMF [34], we then firstly examined the DBU-promoted trifluoromethylthiolation of benzyl bromide (1a) with the difluorocarbene reagent Ph3P+CF2CO2- in the presence of elemental sulfur (S8) (Table 1, entry 1). The desired product was obtained albeit in a low yield, suggesting that Ph3P+CF2CO2- can successfully act as the F- and :CF2 source. A brief survey of the reaction solvents (entries 1–4) revealed that DMA was a superior solvent (entry 2). Besides DBU, various other bases were also screened and some bases were found to be ineffective (entries 5–12). To our delight, 62% yield was obtained by using Cs2CO3 as a base (entry 12). Increasing the loading of elemental sulfur alone led to a slight decrease in the yield (entries 13 and 14 vs. 12). This may be because difluorocarbene would be trapped rapidly by elemental sulfur [38, 39] and the generation of fluoride from difluorocarbene would be slightly suppressed. The yield was also decreased by either decreasing or increasing the loading of Ph3P+CF2CO2- alone (entries 15 and 16 vs. 12). Increasing the loadings of both Ph3P+CF2CO2- and S8 increased the yield to 84% (entry 17).
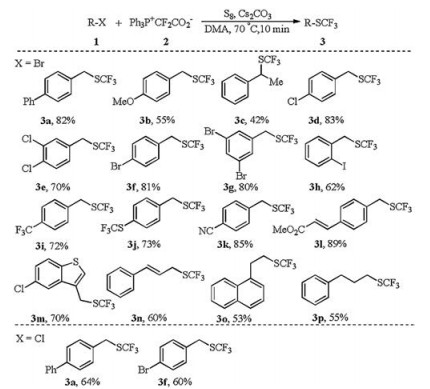
With the optimized reaction conditions in hand (Table 1, entry 17), we then explored the substrate scope of the trifluoromethylthiolation by using Ph3P+CF2CO2- as the fluoride and difluorocarbene source (Scheme 2). Electron-rich, -neutral, and –deficient benzyl bromides could all be well converted into the desired products in moderate to good yields (3a-3m). These conditions could be applied to the conversion of secondary bromides (3c). A low isolated yield of 3c was because of its high volatility (60% 19F NMR yield was obtained before isolation). Besides benzyl bromides, allyl bromides (3n) and alkyl bromides (3o, 3p) also showed good reactivity towards this trifluoromethylthiolation. As shown in Scheme 2, a wide range of functional group could be tolerated, such as aryl halides, cyanide, ester, alkene, and heteroarene. Although the electrophilicity of benzyl chlorides is apparently lower than benzyl bromides, trifluoromethylthiolation of benzyl chlorides proceeded smoothly.

|
Download:
|
| Scheme 2. Substrate scope of trifluoromethylthiolation. Isolated yields. Reaction conditions: substrate 1 (0.2 mmol), Ph3P+CF2CO2- (4 equiv.), S8 (1.25 equiv.), Cs2CO3 (1 equiv.), and DMA (3 mL) at 70 ℃ for 10 min. | |
In this trifluoromethylthiolation process, only -CF2- source and S source were present, and thus the question arose as to how the CF3S moiety was formed in the absence of an external fluoride. It was found that trifluoromethane (HCF3) was rapidly generated as the major product by stirring the mixture of Ph3P+CF2CO2- and Cs2CO3 in DMA (Scheme 3) (details can be found in Supporting information). The low yield (22%) should be because of its low boiling point (-82 ℃). No HCF3 was detected by simply heating Ph3P+CF2CO2- in DMA without the presence of Cs2CO3. The observation of HCF3 meant that the reaction of Ph3P+CF2CO2- with Cs2CO3 produced a fluoride anion, which was trapped by difluorocarbene to form CF3- anion. Although it has been reported that CF3- anion could react with an electrophilic sulfur source to generate CF3S- anion [27, 47], the formation of CF3- anion might not be the predominant path in our reactions. Evidence has been collected in our previous work to reveal that the capture of difluorocarbene by elemental sulfur to give thiocarbonyl fluoride is a very fast process [38, 39]. Besides, as shown in entries 13 and 14 in Table 1, increasing the loading of elemental sulfur alone led to a slight decrease in the yield. This should be because the generation of fluoride anion was suppressed by the more rapid process, the capture of difluorocarbene by elemental sulfur.

|
Download:
|
| Scheme 3. The evidence for the generation of a fluoride anion from difluorocarbene. aThe yield was determined by 19F NMR spectroscopy. | |
On the basis of the above results, we propose the reaction mechanism as shown in Scheme 4. Warming conditions lead to the generation of difluorocarbene from Ph3P+CF2CO2- [31-40]. Difluorocarbeneis electrophilic and would thus beeasily trapped bycesium carbonate to form dianion A. α-Fluoride elimination of intermediate A producescarbene B, whichcollapsestoreleasecarbondioxideand carbonoxide[34], andgeneratesfluorideanion.Therapidcaptureof difluorocarbene byelemental sulfur provides thiocarbonyl fluoride, which is also quite electrophilic and the attack of fluoride at this species gives trifluoromethylthio anion (CF3S-) (Path I). Nucleophilic substitution of a substrate with CF3S- anion furnishes the finalproduct.EventhoughtheformationofCF3-anionanditsattack at elemental sulfur to form CF3S- anion is not the predominant path (Path II), this process may not be excluded.

|
Download:
|
| Scheme 4. The proposed mechanism. | |
In summary, we have described the trifluoromethylthiolation of alkyl halides by using Ph3P+CF2CO2- as a fluoride and difluorocarbene source. The development of a side reaction of difluorocarbene, a process which is usually ignored in organic synthesis, into a synthetic tool may provide foundation for the exploration of difluorocarbene chemistry. The trifluoromethylthiolation process occurred rapidly even though the reaction involved multisequential steps, the cleavage of C–F bond, the formation of CF2=S bond, F-C(S)F2 bond, and C-SCF3 bond. As the difluorocarbene reagent, Ph3P+CF2CO2-, could be easily prepared and is easy to handle, this trifluoromethylthiolation protocol may find application in the synthesis of CF3S-containing biologically active molecules.
AcknowledgmentsWe thank the National Basic Research Program of China (No. 2015CB931903), the National Natural Science Foundation of China (Nos. 21421002, 21472222, 21502214, 21672242, 81273537), the Chinese Academy of Sciences (Nos. XDA02020105, XDA02020106), the Key Research Program of Frontier Sciences (CAS) (No. QYZDJSSW-SLH049), the Key Project of Hunan Provincial Education Department (No. 17A190), the Zhengxiang Scholar Program of the University of South China, Hunan Provincial Hengyang City Joint Fund (No. 2017JJ4050), Hunan Graduate Science and Technology Innovation Projects (No. 2018-400), Program for Innovative Talent Team of Hengyang (No. 2017-1), the Key Project of Hengyang Science and Technology Department (No. 2017KJ166), and Shanghai Research Institute of Chemical Industry Co., LTD. (No. SKL-LCTP-201802) for financial support.
Appendix A. Supplementary dataSupplementarymaterialrelatedtothisarticlecanbefound, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2018.11.013.
|
|
Table 1 The optimization of the reaction conditions.a |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [1] |
A. Leo, C. Hansch, D. Elkins, Chem. Rev. 71 (1971) 525-616. DOI:10.1021/cr60274a001 |
| [2] |
C. Hansch, A. Leo, S.H. Unger, et al., J. Med. Chem. 16 (1973) 1207-1216. DOI:10.1021/jm00269a003 |
| [3] |
C. Hansch, A. Leo, R.W. Taft, Chem. Rev. 91 (1991) 165-195. DOI:10.1021/cr00002a004 |
| [4] |
X.H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 115 (2015) 731-764. DOI:10.1021/cr500193b |
| [5] |
F. Toulgoat, S. Alazet, T. Billard, Eur. J. Org. Chem. 2014 (2014) 2415-2428. DOI:10.1002/ejoc.201301857 |
| [6] |
L. Chu, F.L. Qing, Acc. Chem. Res. 47 (2014) 1513-1522. DOI:10.1021/ar4003202 |
| [7] |
K. Zhang, X. Xu, F. Qing, Chin. J. Org. Chem. 35 (2015) 556-569. DOI:10.6023/cjoc201501017 |
| [8] |
X. Shao, C. Xu, L. Lu, Q. Shen, Acc. Chem. Res. 48 (2015) 1227-1236. DOI:10.1021/acs.accounts.5b00047 |
| [9] |
X. Yang, T. Wu, R.J. Phipps, F.D. Toste, Chem. Rev. 115 (2015) 826-870. DOI:10.1021/cr500277b |
| [10] |
H. Zheng, Y. Huang, Z. Weng, Tetrahedron Lett. 57 (2016) 1397-1409. DOI:10.1016/j.tetlet.2016.02.073 |
| [11] |
S. Rossi, A. Puglisi, L. Raimondi, M. Benaglia, ChemCatChem 10 (2018) 2717-2733. DOI:10.1002/cctc.201800170 |
| [12] |
H. Liu, Z. Gu, X. Jiang, Adv. Synth. Catal. 355 (2013) 617-626. DOI:10.1002/adsc.201200764 |
| [13] |
H. Liu, X. Jiang, Chem. Asian J. 8 (2013) 2546-2563. DOI:10.1002/asia.v8.11 |
| [14] |
P. Zhang, L. Lu, Q. Shen, Acta Chim. Sinica 75 (2017) 744-769. DOI:10.6023/A17050202 |
| [15] |
E.A. Nodiff, S. Lipschutz, P.N. Craig, M. Gordon, J. Org. Chem. 25 (1960) 60-65. DOI:10.1021/jo01071a018 |
| [16] |
J.M. Kremsner, M. Rack, C. Pilger, Oliver Kappe C., Tetrahedron Lett. 50 (2009) 3665-3668. DOI:10.1016/j.tetlet.2009.03.103 |
| [17] |
S. Large, N. Roques, B.R. Langlois, J. Org. Chem. 65 (2000) 8848-8856. DOI:10.1021/jo000150s |
| [18] |
C. Pooput, M. Medebielle, W.R. Dolbier, Org. Lett. 6 (2004) 301-303. DOI:10.1021/ol036303q |
| [19] |
S. Alazet, L. Zimmer, T. Billard, Angew. Chem. Int. Ed. 52 (2013) 10814-10817. DOI:10.1002/anie.201305179 |
| [20] |
X. Shao, X. Wang, T. Yang, L. Lu, Q. Shen, Angew. Chem. Int. Ed. 52 (2013) 3457-3460. DOI:10.1002/anie.v52.12 |
| [21] |
Y.D. Yang, A. Azuma, E. Tokunaga, et al., J. Am. Chem. Soc. 135 (2013) 8782-8785. DOI:10.1021/ja402455f |
| [22] |
R. Pluta, P. Nikolaienko, M. Rueping, Angew. Chem. Int. Ed. 53 (2014) 1650-1653. DOI:10.1002/anie.201307484 |
| [23] |
J.B. Liu, X.H. Xu, Z.H. Chen, F.L. Qing, Angew. Chem. Int. Ed. 54 (2015) 897-900. DOI:10.1002/anie.201409983 |
| [24] |
L.J.C. Bonazaba Milandou, H. Carreyre, S. Alaze, et al., Angew. Chem. Int. Ed. 56 (2017) 169-172. DOI:10.1002/anie.201609574 |
| [25] |
Z. Zhang, Z. Sheng, W. Yu, et al., Nat. Chem. 9 (2017) 970-976. DOI:10.1038/nchem.2789 |
| [26] |
C. Chen, Y. Xie, L. Chu, et al., Angew. Chem. Int. Ed. 51 (2012) 2492-2495. DOI:10.1002/anie.v51.10 |
| [27] |
C. Chen, L. Chu, F.L. Qing, J. Am. Chem. Soc. 134 (2012) 12454-12457. DOI:10.1021/ja305801m |
| [28] |
Y. Huang, X. He, X. Lin, M. Rong, Z. Weng, Org. Lett. 16 (2014) 3284-3287. DOI:10.1021/ol501290p |
| [29] |
D. Brahms, W. Dailey, Chem. Rev. 96 (1996) 1585-1632. DOI:10.1021/cr941141k |
| [30] |
C. Ni, J. Hu, Synthesis 46 (2014) 842-863. DOI:10.1055/s-00000084 |
| [31] |
J. Zheng, J. Cai, J.H. Lin, Y. Guo, J.C. Xiao, Chem. Commun. 49 (2013) 7513-7515. DOI:10.1039/c3cc44271c |
| [32] |
J. Zheng, J.H. Lin, J. Cai, J.C. Xiao, Chem.-Eur. J. 19 (2013) 15261-15266. DOI:10.1002/chem.201303248 |
| [33] |
X.Y. Deng, J.H. Lin, J. Zheng, J.C. Xiao, Chem. Commun. 51 (2015) 8805-8808. DOI:10.1039/C5CC02736E |
| [34] |
J. Zheng, J.H. Lin, X.Y. Deng, J.C. Xiao, Org. Lett. 17 (2015) 532-535. DOI:10.1021/ol503548s |
| [35] |
J. Zheng, J.H. Lin, L.Y. Yu, et al., Org. Lett. 17 (2015) 6150-6153. DOI:10.1021/acs.orglett.5b03159 |
| [36] |
J. Zheng, L. Wang, J.H. Lin, J.C. Xiao, S.H. Liang, Angew. Chem. Int. Ed. 54 (2015) 13236-13240. DOI:10.1002/anie.201505446 |
| [37] |
X.Y. Deng, J.H. Lin, J.C. Xiao, Org. Lett. 18 (2016) 4384-4387. DOI:10.1021/acs.orglett.6b02141 |
| [38] |
J. Zheng, R. Cheng, J.H. Lin, et al., Angew. Chem. Int. Ed. 56 (2017) 3196-3200. DOI:10.1002/anie.201611761 |
| [39] |
J. Yu, J.H. Lin, J.C. Xiao, Angew. Chem. Int. Ed. 56 (2017) 16669-16673. DOI:10.1002/anie.201710186 |
| [40] |
J.J. Luo, M. Zhang, J.H. Lin, J.C. Xiao, J. Org. Chem. 82 (2017) 11206-11211. DOI:10.1021/acs.joc.7b01701 |
| [41] |
C. Zhang, Adv. Synth. Catal. 359 (2017) 372-383. DOI:10.1002/adsc.201601011 |
| [42] |
V.V. Levin, A.L. Trifonov, A.A. Zemtsov, et al., Org. Lett. 16 (2014) 6256-6259. DOI:10.1021/ol503225s |
| [43] |
Y. Qiao, T. Si, M.H. Yang, R.A. Altman, J. Org. Chem. 79 (2014) 7122-7131. DOI:10.1021/jo501289v |
| [44] |
Y. Liu, K. Zhang, Y. Huang, et al., Chem. Commun. 52 (2016) 5969-5972. DOI:10.1039/C6CC00666C |
| [45] |
L.I. Panferova, A.V. Tsymbal, V.V. Levin, M.I. Struchkova, A.D. Dilman, Org. Lett. 18 (2016) 996-999. DOI:10.1021/acs.orglett.6b00117 |
| [46] |
M.Q. Hua, W. Wang, W.H. Liu, et al., J. Fluorine Chem. 181 (2016) 22-29. DOI:10.1016/j.jfluchem.2015.11.003 |
| [47] |
Z. Weng, W. He, C. Chen, et al., Angew. Chem. Int. Ed. 52 (2013) 1548-1552. DOI:10.1002/anie.201208432 |