 2019, Vol. 30
2019, Vol. 30 


b Xingzhi College of Zhejiang Normal University, Jinhua 321004, China
Molecular self-assembly is a ubiquitous phenomenon in nature which plays a vital role not only in achieving specific bio functions but also in the fabrication of functional nanomaterials [1-6]. In recent years, the construction of stimuli-responsive supramolecular self-assembled systems have been actively promoted by chemists and materials scientists [7-9], in particular, a wide variety of smart soft materials have been obtained through the bottom-up self-assembly of small molecules in water [10-13]. In this content, supramolecular hydrogels [14-17] particularly those fabricated from the low-molecular-weight gelators (LMWG) have received of great interest for their far-reaching significance from basic science to a wide range application such as drug delivery or industrial production [18-21]. The dynamic nature of non-covalent interactions makes the supramolecular hydrogels with external stimuli responsive features [16]. Among various low-molecularweight supramolecular hydrogels, those fabricated from photochromophore incorporated hydrogelators are particularly attractive, since the properties and functions of the resulting photoresponsive supramolecular hydrogels can be regulated in a non-invasive and spatiotemporally controllable manner by using light as an external stimulus [22-25]. Up to now, photoresponsive homogeneous or hybrid low-molecular-weight supramolecular hydrogels have already been constructed and investigated extensively [26-29], whereas most of them were based on the hydrogelators possessing an aromatic core and flexible substitutes such as oligopeptides [30-32]. It has been demonstrated that rigid molecules with unusual stiff conformations exhibit a higher assembly tendency to form well-defined 1D structures in the aqueous phase [33, 34]. The gelators with more than one aromatic π-unit (so-called π-gelators) are considered to be more promising candidates in the construction of functional π-gels in the widespread applications from organic electronics to imaging and sensing [35].
Therefore, viable strategies with innovative design of novel hydrogelators with unusual structures might bring in exotic properties and functions of these smart soft materials for advanced applications. In our effort, here we described the fabrication of low-molecular-weight photoresponsive supramolecular hydrogel through a bipyridinium functionalized azobenzene-derived hydrogelator (1) (Scheme 1). The introduction of the cationic aromatic pyridinium modules is expected not only to regulate the solubility of such gelator in aqueous solution, but also provide additional π-π stacking interactions which will significantly facilitate the hydrogel formation. More importantly, benefiting from the reversible E/Z photoisomerization of azobenzene core, which has been widely used in the construction of photoresponsive soft organic materials, the photo-regulated gel-sol transformation could be achieved.

|
Download:
|
| Scheme 1. Chemical structure and the schematic representation of the UV–vis induced E/Z photoisomerization of the azobenzene-derived hydrogelator 1. | |
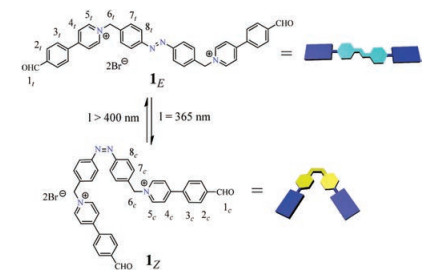{kind=link}
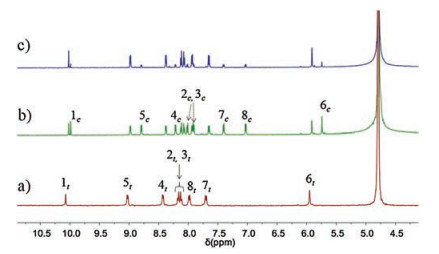
The azobenzene-derived hydrogelator 1 could be conveniently synthesized by following the reported procedures [36] (Scheme S1 in Supporting inforamtion). 1H NMR and UV–vis spectroscopic investigations were first performed to disclose the photoisomerzation behaviors of 1 in water. As displayed in Fig. 1, when the solution of 1 was irradiated by the UV light of λ = 365 nm for 1.0 h to reach the photostationary state (PSSZ), a new set of well-resolved proton signals (1c - 8c in Fig. 1b) corresponding to 1Z appeared. This suggested that the E→Z photoisomerization of hydrogelator 1 has been successfully triggered in the solution. After irradiating with the

|
Download:
|
| Fig. 1. Partial 1H NMR spectra (600 MHz, D2O, 2.0 mmol/L, 25 ℃) of 1 (a) before and (b) after irradiation with UV light (λ = 365 nm, 35 mW/cm2) for 1.0 h at 25 ℃; (c) the UV-irradiated 1 after irradiation with visible light (λ > 400 nm, 100 mW/cm2) for 1.0 h at 25 ℃. | |
{kind=link}
visible light of λ > 400 nm for another 1.0 h, this PSSZ mixtures of 1 could be further converted to the PSSE mixtures as proved by the observation of dramatically decreased proton signals of 1Z (Fig. 1c), which means the Z-isomer has been converted back to the E-isomer. By integrating the proton signals of 6c/6t, the ratio of the two isomers in the PSSZ mixtures of 1 could be estimated as 1Z (60%) and 1E (40%), which turn out to be about 1Z (20%) and 1E (80%) in the PSSE mixtures. The multicomponent PSSs of 1 could be attributed to the incomplete photoisomerzation processes in both directions.
UV-vis spectra were further recorded to reveal the photoisomerization behaviors of 1 in water. Before irradiation, the solution of 1 exhibited a strong absorption band at 250–375 nm (black line in Fig. 2a) corresponding to the combined absorption of the terminal cationic pyridinium substituents and the π-π* absorbance of azobenzene core. The n-π* absorption band of the azobenzene unit appeared at ca. 430 nm which was relative weak and broad (black line in Fig. 2b). Upon the irradiation by UV light (λ = 365 nm) to reach PSSZ, during which 1E could be photoisomerized to 1Z as supported by the dramatically decreasing of the π-π* absorbance at about 330 nm (red line in Fig. 2a) and the increasing of n-π* band (red line in Fig. 2b), meanwhile both absorption bands were slightly blue-shifted. When the solution of the PSSZ mixtures of 1 was exposed to the visible light (λ > 400 nm) to achieved PSSE, the E→Z photoisomerization could be triggered, resulting in the enhanced π-π* and decreased n-π* absorption bands (blue line in Fig. 2).

|
Download:
|
| Fig. 2. (a) The full UV–vis absorption spectra of 1 (0.040 mmol/L) and (b) partial UV–vis absorption spectra of 1 (0.25 mmol/L) in H2O at 25 ℃. Black line: before irradiation; red line: after irradiation with UV light (λ = 365 nm, 35 mW/cm2) for 0.5 h; and blue line: the UV-irradiated solution of 1 after irradiation by visible light (λ > 400 nm, 100 mW/cm2) for another 0.5 h. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article). | |
{kind=link}
After revealing the UV–vis light induced E/Z photoisomerization behaviors of 1 in the aqueous solution, we turn our attention to the self-assembly properties of 1 under different irradiation conditions. Before irradiation, hydrogel that can stand upsidedown was formed as the concentration of non-irradiated 1 increased up to 5 mmol/L in water at 25 ℃ (left picture in Fig. 3a), which could be converted to the solution phase after irradiating with UV light (λ = 365 nm) (right picture in Fig. 3a). When the UV-irradiated solution of 1 was exposed to the visible light (λ > 400 nm), the hydrogel could be regenerated. Therefore, reversible light-regulated gel-sol transformation of 1 could be achieved by employing alternating UV–vis light irradiations. The morphologies of 1 at different PSSs were further revealed by the scanning electron microscopy (SEM) experiments, from which well-defined fibers formed for 1 at PSSE (Fig. 3b) but disordered assemblies for 1 at PSSZ (Fig. 3c) could be observed. The formation of one-dimensional ordered fibers might be attributed to the combined non-covalent interactions of 1E including the intermolecular π-π stacking between the aromatic segments as well as the hydrophobic interactions with water, which is analogous to selfassembly mechanisms of other π-gelators in water [35]. However, the UV-light irradiation will trigger the E→Z photoisomerization of hydrogelator 1, which is likely to convert the planar E-isomer to the twisted Z-isomer. The large steric repulsion between the Z-isomers is likely to weak the intermolecular π-π stacking interactions, which will lead to the destruction of the fibrous architectures. Furthermore, it is worth mentioning that the reversible gel-sol transformation of this supramolecular hydrogel could also be achieved by the alternating heating/cooling treatments of the aqueous solution of non-irradiated 1.

|
Download:
|
| Fig. 3. (a) Pictures of the reversible gel-sol transformation of supramolecular hydrogel fabricated from the solution of 1 (5.0 mmol/L in H2O) upon alternating UV–vis light irradiation at 25 ℃. (b) SEM images of the samples prepared from the aqueous solution of 1 (0.25 mmol/L) before and (c) after irradiation with UV light (λ = 365 nm) for 0.5 h at 25 ℃. The scare bar of SEM images is 1.0μm. | |
{kind=link}
In order to get more insight into the self-assembly mechanism of 1 in water, the powder X-ray diffraction (PXRD) data were further collected. As displayed in Fig. 4, the PXRD profile of the non-irradiated dried hydrogel sample exhibited a strong diffraction peak at approximately 2θ = 25° (black line), indicating the existence of strong aromatic stacking interactions between the 1E molecules in the gel formation. On the contract, the UV-irradiated sample revealed relatively weak diffraction signal at this region (red line), which suggested there was short of efficient stacking interactions in these ill-defined assemblies. Based on these combined observations, the gelation mechanism could be proposed as that the planar E-isomer of hydrogelator 1 was more favored to generate strong π-π stacking interactions, resulting in the formation of well-defined 1D fibrous assemblies, which could be further intertwined to form 3D networks [35]. It was worth mentioning that the existence of two terminal aldehyde groups could make positive contribution on the gelation of 1E by extending the aromatic conjugations, besides, further functionalization of such hydrogelator could be expected by the reaction of these aldehyde groups.

|
Download:
|
| Fig. 4. Powder XRD patterns of the samples prepared from 1 before (black) and after (red) irradiation by UV light (λ = 365 nm) for 1.0 h at 25 ℃. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article). | |
{kind=link}
In summary, we have demonstrated that low-molecular-weight photoresponsive supramolecular hydrogel could be fabricated from a simple dicationic azobenzene-bridged pyridinium salt in the aqueous solution. Thanks to the reversible photoswitchability of the incorporated azobenzene photochromophore, the resulting supramolecular hydrogel exhibits interesting photo-induced reversible gel-sol transformation. The introduction of the cationic aromatic pyridinium substituents to the photochromophore core has been proved to be an accessible strategy in the fabrication of photoresponsive low-molecular-weight supramolecular hydrogel. This strategy may serve as alternative innovative bottom up approach in the rational molecular design of smart functional soft materials with predictable behaviors and properties.
AcknowledgmentsThe authors are grateful to the National Natural Science Foundation of China (Nos. 21502216 and 21602205) and Natural Science Foundation of Zhejiang Province (No. LQ19B020019) for the financial support to this research. They also appreciate Prof. Xin Zhao at Shanghai Institute of Organic Chemistry (SIOC), CAS, for the helpful discussions.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.10.024.
| [1] |
G.M. Whitesides, B. Grzybowski, Science 295 (2002) 2418-2421. DOI:10.1126/science.1070821 |
| [2] |
C. Wang, Z. Wang, X. Zhang, Acc. Chem. Res. 45 (2012) 608-618. DOI:10.1021/ar200226d |
| [3] |
D.S. Guo, Y. Liu, Chem. Soc. Rev. 41 (2012) 5907-5921. DOI:10.1039/c2cs35075k |
| [4] |
Y. Kim, W. Li, S. Shin, M. Lee, Acc. Chem. Res. 46 (2013) 2888-2897. DOI:10.1021/ar400027c |
| [5] |
S.I. Stupp, L.C. Palmer, Chem. Mater. 26 (2014) 507-518. DOI:10.1021/cm403028b |
| [6] |
M. Liu, L. Zhang, T. Wang, Chem. Rev. 115 (2015) 7304-7397. DOI:10.1021/cr500671p |
| [7] |
X. Yan, F. Wang, B. Zheng, F. Huang, Chem. Soc. Rev. 41 (2012) 6042-6065. DOI:10.1039/c2cs35091b |
| [8] |
X. Ma, H. Tian, Acc. Chem. Res. 47 (2014) 1971-1981. DOI:10.1021/ar500033n |
| [9] |
X.Y. Hu, T. Xiao, C. Lin, F. Huang, L. Wang, Acc. Chem. Res. 47 (2014) 2041-2051. DOI:10.1021/ar5000709 |
| [10] |
J. Tian, H. Wang, D.W. Zhang, Y. Liu, Z.T. Li, Sci. Rev. 4 (2017) 426-436. |
| [11] |
G. Wu, J. Thomas, M. Smet, Z. Wang, X. Zhang, Chem. Sci. 5 (2014) 3267-3274. DOI:10.1039/C4SC00860J |
| [12] |
van Dijken D.J., J. Chen, M.C.A. Stuart, L. Hou, B.L. Feringa, J. Am. Chem. Soc. 138 (2016) 660-669. DOI:10.1021/jacs.5b11318 |
| [13] |
Q. Zhao, Y. Chen, Y. Liu, Chin. Chem. Lett. 29 (2018) 84-86. DOI:10.1016/j.cclet.2017.07.024 |
| [14] |
E.A. Appel, del Barrio J., X.J. Loh, O.A. Scherman, Chem. Soc. Rev. 41 (2012) 6195-6214. DOI:10.1039/c2cs35264h |
| [15] |
R.G. Weiss, J. Am. Chem. Soc. 136 (2014) 7519-7530. DOI:10.1021/ja503363v |
| [16] |
X. Du, J. Zhou, J. Shi, B. Xu, Chem. Rev. 115 (2015) 13165-13307. DOI:10.1021/acs.chemrev.5b00299 |
| [17] |
W. Zheng, L.J. Chen, G. Yang, et al., J. Am. Chem. Soc. 138 (2016) 4927-4937. DOI:10.1021/jacs.6b01089 |
| [18] |
L.A. Estroff, A.D. Hamilton, Chem. Rev. 104 (2004) 1201-1217. DOI:10.1021/cr0302049 |
| [19] |
Z. Yang, G. Liang, B. Xu, Acc. Chem. Res. 41 (2008) 315-326. DOI:10.1021/ar7001914 |
| [20] |
M. Suzuki, K. Hanabusa, Chem. Soc. Rev. 38 (2009) 967-975. DOI:10.1039/b816192e |
| [21] |
E.R. Draper, D.J. Adams, Chemistry 3 (2017) 390-410. DOI:10.1016/j.chempr.2017.07.012 |
| [22] |
I. Tomatsu, A. Hashidzume, A. Harada, Macromolecules 38 (2005) 5223-5227. DOI:10.1021/ma050670v |
| [23] |
Y.L. Zhao, J.F. Stoddart, Langmuir 25 (2009) 8442-8446. DOI:10.1021/la804316u |
| [24] |
D. Wang, M. Wagner, H.J. Butt, Wu Si, Soft Matter 11 (2015) 7656-7662. DOI:10.1039/C5SM01888A |
| [25] |
X. Yao, T. Li, J. Wang, X. Ma, H. Tian, Adv. Optical Mater. 4 (2016) 1322-1349. DOI:10.1002/adom.201600281 |
| [26] |
E.R. Draper, D.J. Adams, Chem. Commun. 52 (2016) 8196-8206. DOI:10.1039/C6CC03485C |
| [27] |
W. Fang, X. Liu, Z. Lu, T. Tu, Chem. Commun. 50 (2014) 3313-3316. DOI:10.1039/C3CC49402K |
| [28] |
Z. Li, G. Wang, Y. Wang, H. Li, Angew. Chem. Int. Ed. 57 (2018) 2194-2198. DOI:10.1002/anie.v57.8 |
| [29] |
L. Ji, G. Ouyang, M. Liu, Langmuir 33 (2017) 12419-12426. DOI:10.1021/acs.langmuir.7b02285 |
| [30] |
H. Kobayashi, A. Friggeri, K. Koumoto, et al., Org. Lett. 4 (2002) 1423-1426. DOI:10.1021/ol025519+ |
| [31] |
D. Wu, X. Xie, A.A. Kadi, Y. Zhang, Chin. Chem. Lett. 29 (2018) 1098-1104. DOI:10.1016/j.cclet.2018.04.030 |
| [32] |
T. Bhattacharyya, P. Saha, J. Dash, ACS Omega 3 (2018) 2230-2241. DOI:10.1021/acsomega.7b02039 |
| [33] |
F. Lin, T.Y. Zhou, T.G. Zhan, X. Zhao, Tetrahedron 70 (2014) 2251-2256. DOI:10.1016/j.tet.2014.02.029 |
| [34] |
F. Lin, R. Liang, Q. Qi, et al., Chin. J. Chem. 35 (2017) 429-434. DOI:10.1002/cjoc.v35.4 |
| [35] |
S.S. Babu, V.K. Praveen, A. Ajayaghosh, Chem. Rev. 114 (2014) 1973-2129. DOI:10.1021/cr400195e |
| [36] |
L. Zhu, M. Lu, D. Qu, Q. Wang, H. Tian, Org. Biomol. Chem. 9 (2011) 4226-4233. DOI:10.1039/c0ob01124j |